Genetic-1-B.sc-unit-2-Special Pathology Pathological changes in disease conditions of selected systems
Special Pathology Pathological changes in disease conditions of selected systems:
Respiratory system
Pathological Changes in Pneumonia
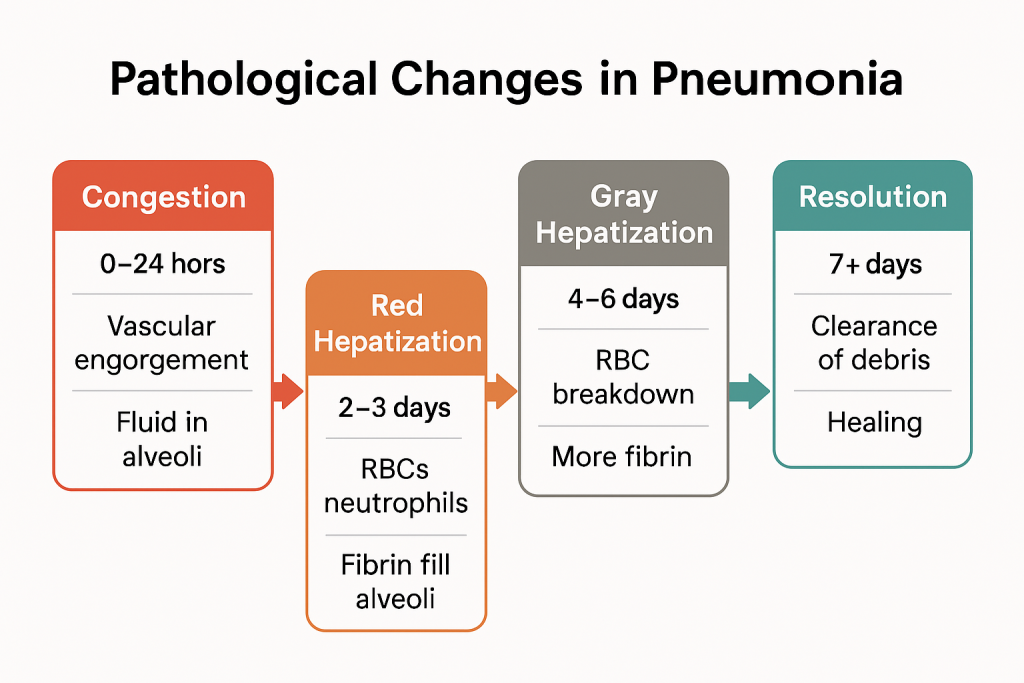
Pneumonia is an inflammatory disease of the lung parenchyma, primarily affecting the alveoli and interstitial tissue. It is typically caused by infectious agents (bacteria, viruses, fungi) and leads to a series of progressive pathological changes in the lungs.
These pathological changes can be best understood in four classic stages, especially in lobar pneumonia, a prototype form of the disease.
1. Congestion (Initial Stage)
🕒 Timeframe: 0–24 hours
- Pathology:
- The alveolar capillaries become engorged with blood due to inflammation.
- There is exudation of protein-rich fluid into the alveoli.
- Numerous bacteria proliferate in the alveolar spaces.
- The alveoli begin to fill with serous fluid, few neutrophils, and scattered red blood cells (RBCs).
- Gross Appearance:
- The lung appears heavy, dark red, and boggy.
- Microscopy:
- Dilated capillaries, early neutrophilic infiltration, and fluid-filled alveoli.
2. Red Hepatization (Consolidation Stage)
🕒 Timeframe: 2–3 days
- Pathology:
- Massive exudation of RBCs, neutrophils, and fibrin into alveolar spaces.
- The lung becomes solidified—similar in appearance and consistency to the liver (hence “hepatization”).
- Gross Appearance:
- Lung is red, firm, and airless.
- Microscopy:
- Alveoli are packed with RBCs, neutrophils, and fibrin strands.
- Alveolar septa may show congestion and inflammatory infiltration.
3. Gray Hepatization (Late Consolidation)
🕒 Timeframe: 4–6 days
- Pathology:
- RBCs within the alveoli disintegrate.
- Neutrophils persist, and fibrin continues to accumulate.
- Alveolar spaces are filled with fibrinopurulent exudate.
- The lung loses its red color and becomes grayish.
- Gross Appearance:
- Lung is gray, dry, and firm.
- Microscopy:
- Fewer RBCs, heavy neutrophilic infiltration, and fibrin deposition.
- Septal thickening may occur due to inflammation.
4. Resolution (Healing Stage)
🕒 Timeframe: 7–10 days onward
- Pathology:
- Macrophages digest and remove the fibrin and necrotic debris.
- Exudate is cleared via cough, mucociliary action, or lymphatics.
- Lung architecture is gradually restored to normal, assuming no complications arise.
- Gross Appearance:
- Lung becomes soft and pink again.
- Microscopy:
- Clearing of alveolar spaces, regeneration of epithelial cells, and reduced inflammatory cells.
Summary in Chart Form
| Stage | Timeframe | Key Features | Gross Appearance | Microscopic Findings |
|---|---|---|---|---|
| Congestion | 0–24 hrs | Vascular engorgement, fluid in alveoli | Red, boggy, heavy | Dilated capillaries, few neutrophils |
| Red Hepatization | 2–3 days | RBCs, neutrophils, fibrin fill alveoli | Red, solid, liver-like | Dense exudate in alveoli |
| Gray Hepatization | 4–6 days | RBC breakdown, more fibrin | Gray, dry, firm | Fibrin, neutrophils, less RBCs |
| Resolution | 7+ days | Clearance of debris, healing | Soft, pink | Alveoli cleared, macrophages dominate |
Clinical Relevance
- Understanding these stages is essential for timing of clinical intervention, anticipating complications like abscess, pleural effusion, or fibrosis, and interpreting radiological or histological findings.
- In atypical or viral pneumonia, these stages may be incomplete or absent, and the pathological process involves more interstitial inflammation than alveolar filling.
Pathological Changes in Lung Abscess
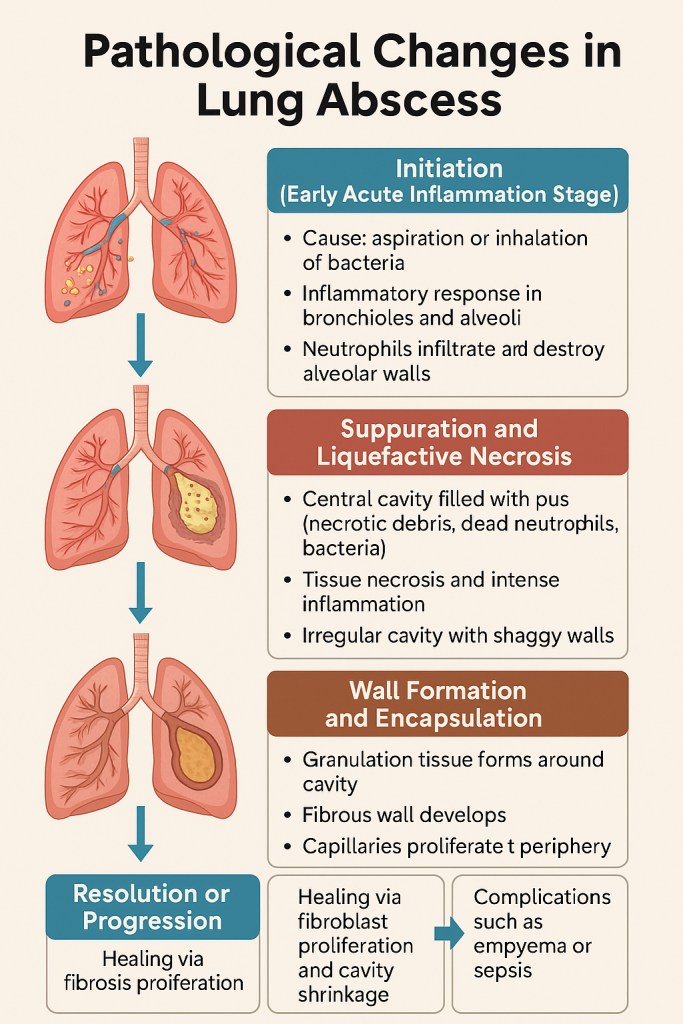
A lung abscess is a localized area of suppurative necrosis within the pulmonary parenchyma resulting in cavity formation, typically due to microbial infection.
It represents a severe inflammatory response and tissue destruction, often following aspiration pneumonia, septic embolism, or bronchial obstruction. The pathological process evolves in distinct stages from infection to fibrosis.
1. Initiation (Early Acute Inflammation Stage)
- Cause: Inhalation or aspiration of pathogenic organisms (commonly anaerobic bacteria, Klebsiella, Staphylococcus aureus).
- Pathology:
- Inflammatory response begins in the bronchioles and alveoli.
- Neutrophils infiltrate and destroy alveolar walls.
- Intense vasodilation, edema, and congestion of the surrounding tissue occur.
- Tissue Appearance: Swollen, congested, with microabscesses forming around bronchioles.
2. Suppuration and Liquefactive Necrosis
- Pathology:
- Tissue necrosis progresses rapidly.
- A central cavity forms, filled with pus (dead neutrophils, bacteria, necrotic debris).
- Surrounding tissue shows intense inflammatory infiltration.
- Cavity:
- The abscess cavity may be single or multiple, irregular, with shaggy walls.
- Can communicate with bronchi, leading to expectoration of foul-smelling sputum.
- Gross Appearance:
- Yellow-gray cavity with purulent exudate, surrounded by inflamed and necrotic lung.
3. Wall Formation and Encapsulation
- Pathology:
- As healing begins, granulation tissue forms around the cavity.
- Over time, this leads to fibrous encapsulation.
- Capillaries proliferate at the periphery, trying to isolate the infection.
- Clinical Significance:
- This stage determines whether the abscess will resolve, persist, or progress to complications.
4. Resolution or Progression
- Resolution:
- If the abscess drains properly into the bronchus, healing may occur.
- Fibroblast proliferation leads to scar formation and cavity shrinkage.
- Progression/Complications:
- If drainage fails or the infection spreads, complications such as:
- Bronchopleural fistula
- Empyema (pus in pleural space)
- Septicemia
- Chronic lung abscess may occur.
- If drainage fails or the infection spreads, complications such as:
Summary in Chart Form
| Stage | Key Events & Features |
|---|---|
| Initiation (Acute Inflammation) | Infiltration by neutrophils, alveolar congestion, microabscess formation |
| Suppuration & Necrosis | Cavity formation, filled with pus, necrotic lung parenchyma |
| Wall Formation & Encapsulation | Granulation tissue develops, fibrous wall surrounds cavity |
| Resolution or Progression | Healing via fibrosis or complications like empyema, sepsis |
Clinical Relevance for Nursing & Medicine:
- Monitoring sputum color and odor gives clues about cavity communication.
- Chest X-ray or CT scan often reveals air-fluid levels within the abscess.
- Early identification and antibiotic therapy can prevent fibrosis and complications.
- Nursing roles include airway clearance, postural drainage, and infection control.
Pathological Changes in Pulmonary Tuberculosis
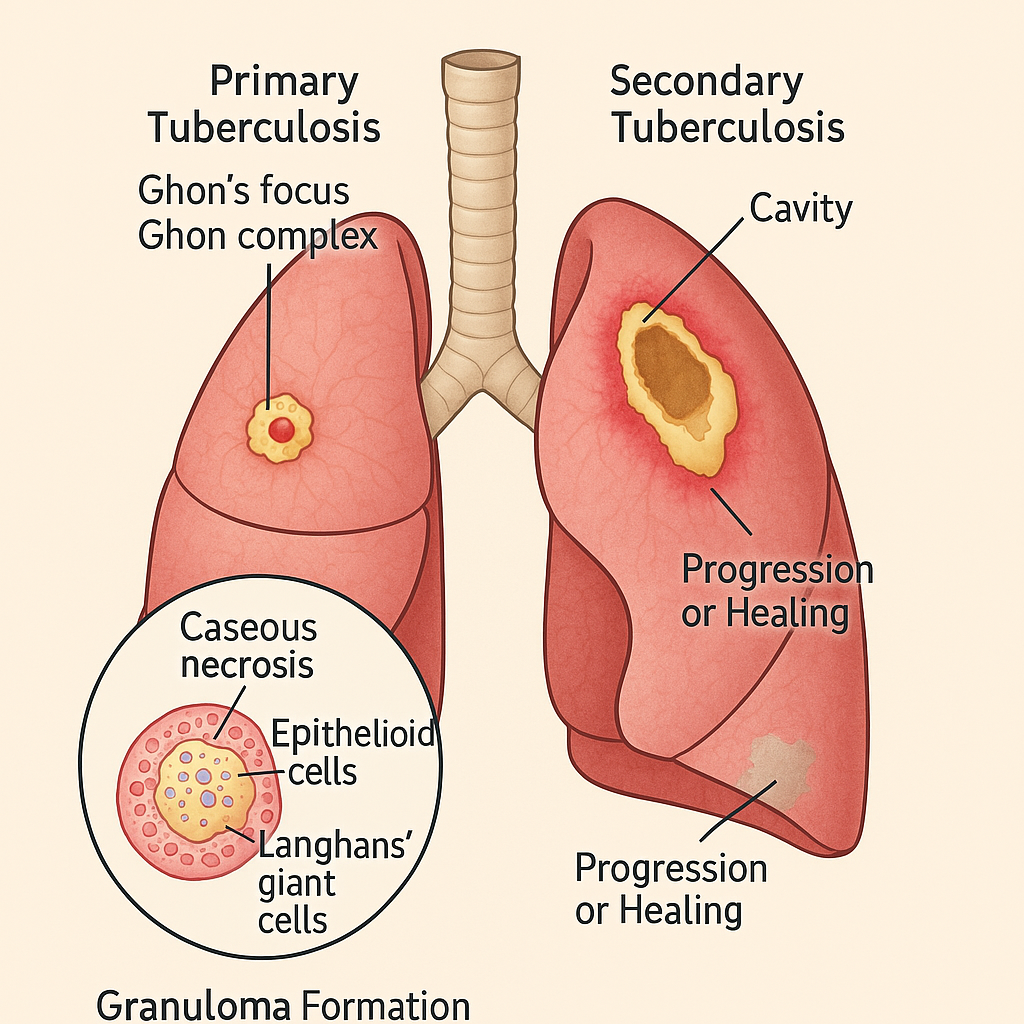
Pulmonary tuberculosis is a chronic granulomatous infection of the lung caused by Mycobacterium tuberculosis. The hallmark of TB is the formation of caseating granulomas and progressive tissue necrosis.
The pathological changes in TB are classified based on:
- Type of infection: Primary vs. Secondary TB
- Immune response: Cell-mediated immunity (CMI)
- Disease stage: Initial infection → Granuloma formation → Caseation → Cavitation → Healing or progression
1. Primary Tuberculosis (Ghon’s Complex Formation)
🕒 Seen in: First-time exposure, mostly children
- Pathology:
- Bacilli enter alveoli → engulfed by alveolar macrophages but not destroyed.
- Localized inflammatory response → formation of a small subpleural granuloma, often in the mid-lung zone.
- Infection spreads to regional lymph nodes → lymphadenopathy.
- Ghon’s Complex:
- Triad of:
- Parenchymal lesion (Ghon focus)
- Involved hilar lymph nodes
- Lymphangitis
- Triad of:
- Microscopy:
- Central caseation necrosis, epithelioid cells, Langhans’ giant cells, surrounding lymphocytes.
- Fate:
- Heals by fibrosis and calcification, forming a Ranke complex.
2. Secondary (Post-primary) Tuberculosis
🕒 Seen in: Reactivation or reinfection, often in adults
- Site: Apex of lungs (high oxygen tension favors bacilli)
- Pathology:
- Intense hypersensitivity reaction (Type IV), leading to:
- Caseous necrosis
- Liquefaction
- Cavity formation
- Intense hypersensitivity reaction (Type IV), leading to:
- Cavity Formation:
- Caseous debris liquefies and drains via bronchi → cavity with ragged walls.
- These cavitations are highly infectious, shedding bacilli into the airways.
- Surrounding Changes:
- Extensive fibrosis
- Adjacent lung destruction
- Bronchiectasis
- Pleural thickening or effusion
3. Granuloma Formation (Tubercle)
- Structure of a Tuberculous Granuloma:
- Central caseous necrosis
- Ring of epithelioid cells
- Langhans’ giant cells
- Outer layer of lymphocytes and fibroblasts
- Function:
- Attempt to contain infection.
- Poor vascularization leads to hypoxia → caseation.
4. Progression or Healing
- Healing:
- Caseous foci may be replaced by:
- Fibrosis
- Calcification
- Scar tissue
- Caseous foci may be replaced by:
- Progression/Complications:
- Miliary TB: Dissemination via bloodstream (tiny millet seed-like lesions in lungs)
- Pleural effusion, Empyema
- Bronchopleural fistula
- Massive hemoptysis
- Post-tubercular fibrosis and collapse
Summary in Chart Form
| Stage/Type | Key Pathological Features |
|---|---|
| Primary TB | Ghon complex (lung + lymph nodes), caseation, calcification |
| Secondary TB | Apical lesions, caseation, cavitation, fibrosis |
| Granuloma (Tubercle) | Central caseation, epithelioid cells, Langhans’ giant cells |
| Healing | Fibrosis, calcified scar, possible cavity closure |
| Complications | Miliary spread, pleural involvement, hemoptysis, bronchiectasis |
Clinical and Nursing Relevance
- Persistent cough, hemoptysis, weight loss, and night sweats are key indicators.
- Cavitary lesions in the apex on X-ray suggest secondary TB.
- Sputum AFB test and Mantoux test aid in diagnosis.
- Nurses play a vital role in infection control, DOTS compliance, nutrition, and psychosocial support.
Pathological Changes in Chronic Bronchitis
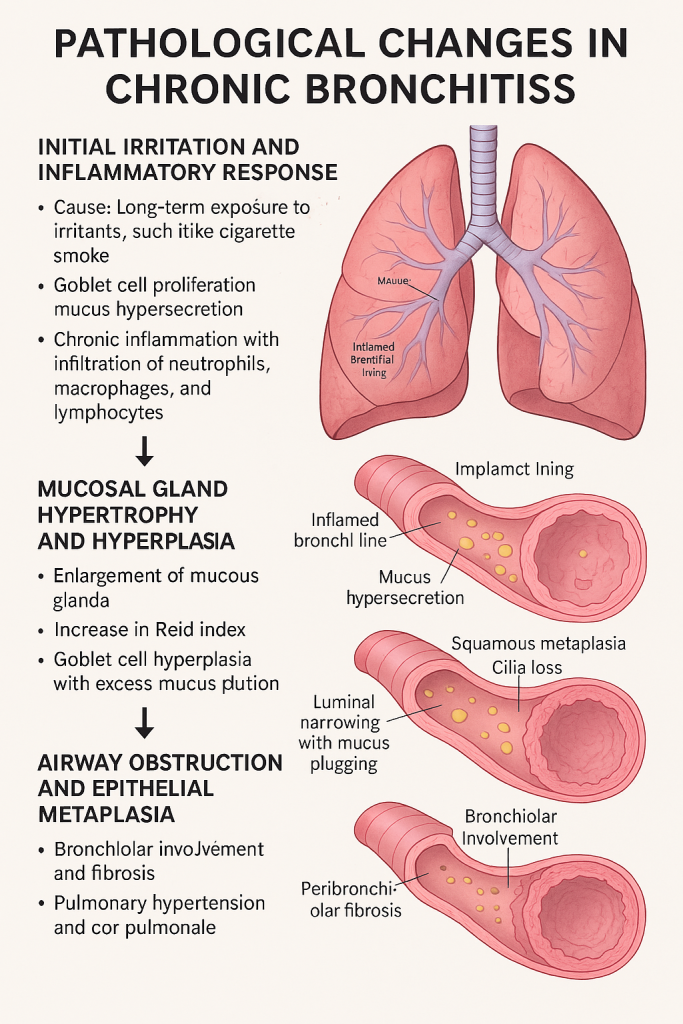
Chronic bronchitis is a type of chronic obstructive pulmonary disease (COPD) characterized by persistent inflammation of the bronchi, leading to mucus hypersecretion, airway obstruction, and progressive lung damage.
Definition:
Chronic bronchitis is clinically defined as a productive cough lasting at least 3 months in 2 consecutive years, not due to other causes.
1. Initial Irritation and Inflammatory Response
- Cause: Long-term exposure to irritants, especially cigarette smoke, air pollution, dust, or occupational fumes.
- Pathology:
- The bronchial epithelium is exposed to irritants.
- Goblet cells proliferate and produce excess mucus.
- Chronic inflammation ensues with infiltration of neutrophils, macrophages, and lymphocytes.
- Damage begins in the large bronchi, especially centrally.
- Early symptoms: Productive cough, increased mucus.
2. Mucosal Gland Hypertrophy and Hyperplasia
- Pathology:
- Mucous glands in the submucosa enlarge.
- Increase in Reid Index (ratio of gland thickness to wall thickness; normal <0.4, raised in CB).
- Goblet cell hyperplasia further increases mucus production.
- Effect:
- Air passages become clogged with thick mucus, impairing airflow.
- Cilia become damaged → reduced mucociliary clearance.
3. Airway Obstruction and Epithelial Metaplasia
- Chronic damage to the bronchial lining leads to:
- Squamous metaplasia (normal pseudostratified columnar epithelium replaced by squamous cells).
- Loss of cilia, impairing defense mechanisms.
- Persistent airway narrowing due to edema, inflammation, and fibrosis.
- Clinically:
- Dyspnea on exertion.
- Morning cough with copious sputum.
4. Bronchiolar Involvement and Fibrosis
- Small airways (bronchioles) also get involved over time.
- Bronchiolitis obliterans may occur due to fibrotic narrowing.
- Mucus plugging worsens with peribronchiolar fibrosis.
- V/Q mismatch arises → hypoxemia.
5. Pulmonary Hypertension and Cor Pulmonale
- Long-standing hypoxia leads to:
- Vasoconstriction of pulmonary arteries → pulmonary hypertension.
- Right ventricular hypertrophy (RVH) → cor pulmonale (right heart failure due to lung disease).
- Peripheral edema, cyanosis, hepatomegaly can follow.
Summary in Chart Form
| Stage/Feature | Key Pathological Changes |
|---|---|
| Initial Inflammation | Goblet cell hyperplasia, inflammatory infiltrate |
| Mucosal Gland Hyperplasia | Increased mucus, raised Reid index |
| Epithelial Changes | Squamous metaplasia, cilia loss |
| Bronchiolar Obstruction | Small airway fibrosis, mucus plugging |
| Pulmonary Vascular Changes | Hypoxic vasoconstriction → pulmonary hypertension, cor pulmonale |
Clinical and Nursing Relevance
- Blue Bloater: A classical term for chronic bronchitis patients due to cyanosis and obesity.
- Complications: Recurrent infections, cor pulmonale, respiratory failure.
- Nursing roles:
- Encourage smoking cessation, breathing exercises.
- Educate about airway clearance techniques.
- Monitor oxygen therapy carefully to avoid CO₂ retention.
Pathological Changes in Emphysema
Emphysema is a chronic, progressive pulmonary disorder classified under Chronic Obstructive Pulmonary Disease (COPD). It is characterized by abnormal permanent enlargement of airspaces distal to the terminal bronchioles, accompanied by destruction of alveolar walls without obvious fibrosis.
Key Focus:
- Destruction of alveolar walls
- Loss of elastic recoil
- Air trapping
- Progressive breathlessness
1. Initial Irritant Exposure and Inflammatory Response
- Main Cause: Cigarette smoking, air pollutants, or alpha-1 antitrypsin deficiency.
- Pathogenesis begins with:
- Inhaled irritants triggering chronic inflammation.
- Recruitment of neutrophils, macrophages, and CD8+ T-lymphocytes.
- These cells release proteolytic enzymes like elastase and matrix metalloproteinases (MMPs).
- Imbalance Between:
- Proteases (destructive enzymes) and
- Anti-proteases (protective enzymes like alpha-1 antitrypsin)
2. Alveolar Wall Destruction and Airspace Enlargement
- Pathology:
- Elastin in alveolar walls is broken down.
- Alveoli lose their septae, resulting in larger, less efficient airspaces.
- Pulmonary capillary beds are also destroyed → ↓ gas exchange surface area.
- Gross Morphology:
- Lungs appear large, overdistended, and pale.
- Blebs or bullae (air-filled spaces >1 cm) may form near the pleura.
- Microscopy:
- Thinned alveolar walls.
- Merged airspaces with loss of architecture.
3. Loss of Elastic Recoil and Air Trapping
- Mechanism:
- Loss of elastic tissue reduces the lungs’ ability to recoil during expiration.
- Small airways collapse prematurely, especially during forced expiration.
- Air becomes trapped → leading to hyperinflation.
- Clinical Sign:
- Prolonged expiration, barrel chest, use of accessory muscles.
4. Pulmonary Vascular Changes and Complications
- Reduced alveolar-capillary interface → ↓ oxygen uptake.
- Hypoxia-induced vasoconstriction in pulmonary arteries → pulmonary hypertension.
- May lead to right-sided heart failure (cor pulmonale).
- Additional Complications:
- Pneumothorax (rupture of bullae)
- Recurrent respiratory infections
- Progressive hypoxia and respiratory failure
Types of Emphysema (Optional Academic Note)
| Type | Area Affected | Common Cause |
|---|---|---|
| Centrilobular | Central part of acinus (respiratory bronchioles) | Smoking (upper lobes) |
| Panacinar | Entire acinus uniformly | Alpha-1 antitrypsin deficiency (lower lobes) |
| Paraseptal | Distal acinus near pleura | Bullae formation, spontaneous pneumothorax |
| Irregular | Scarring from inflammation | Asymptomatic, incidental |
Summary in Chart Form
| Stage / Change | Key Features |
|---|---|
| Inflammatory Initiation | Neutrophils, macrophages release elastase, MMPs |
| Alveolar Wall Destruction | Loss of septae, enlarged airspaces, ↓ surface area |
| Loss of Elastic Recoil | Air trapping, hyperinflation, barrel chest |
| Pulmonary Vascular Changes | Capillary destruction → hypoxia → pulmonary hypertension |
| Complications | Pneumothorax, infections, cor pulmonale |
Clinical and Nursing Relevance
- Pink Puffer: Term used for emphysema-dominant COPD patients—thin, pursed-lip breathing, relatively well oxygenated.
- Nursing care focuses on:
- Oxygen therapy (low-flow)
- Pulmonary rehab (breathing exercises)
- Infection prevention, nutritional support, and patient education
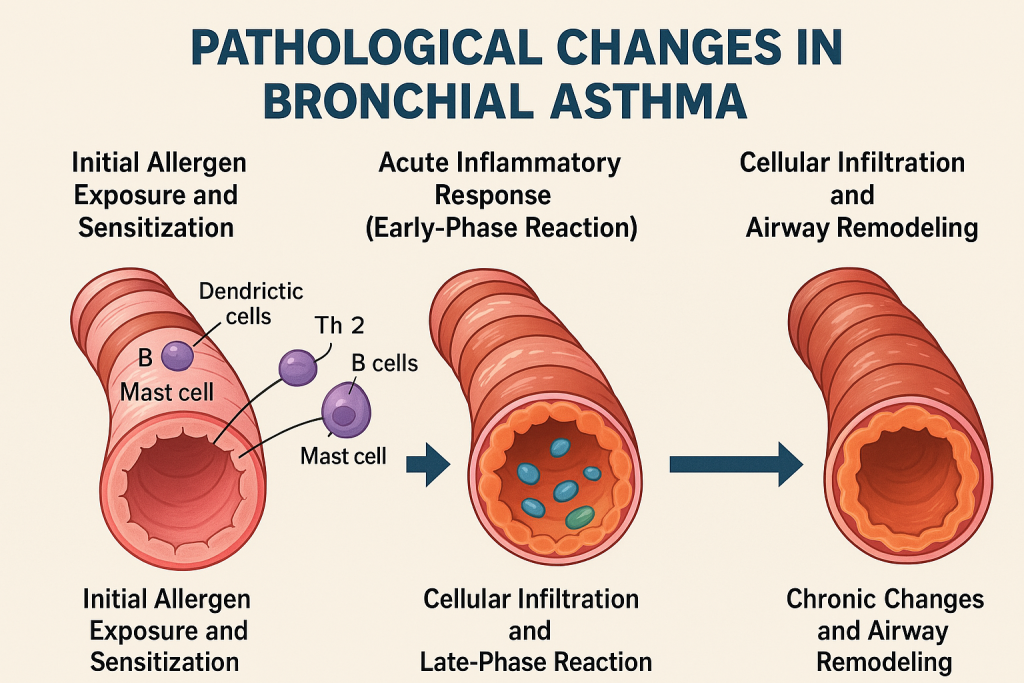
Pathological Changes in Bronchial Asthma
Bronchial asthma is a chronic inflammatory disorder of the airways characterized by:
- Reversible bronchoconstriction,
- Airway hyperresponsiveness, and
- Airway remodeling in long-standing disease.
Asthma involves an exaggerated response to triggers like allergens, infections, cold air, exercise, or irritants. The hallmark feature is intermittent narrowing of the bronchi and bronchioles due to a complex immune and cellular process.
1. Initial Allergen Exposure and Sensitization
- In atopic asthma, exposure to allergens (dust mites, pollen, animal dander) activates:
- Dendritic cells → present allergens to naïve T-cells.
- Differentiation into Th2 helper T-cells → release IL-4, IL-5, IL-13.
- These cytokines stimulate:
- IgE production (via B-cells),
- Eosinophil recruitment, and
- Mast cell sensitization.
- The individual becomes “primed” for hypersensitive response on future exposures.
2. Acute Inflammatory Response (Early-Phase Reaction)
- Upon re-exposure, the allergen cross-links IgE on mast cells → degranulation.
- Release of:
- Histamine: causes bronchoconstriction and vasodilation.
- Leukotrienes (LTC4, LTD4): potent bronchoconstrictors.
- Prostaglandins: promote inflammation.
- Bronchial smooth muscle contraction, edema of airway wall, and mucus hypersecretion result in acute airway obstruction.
3. Cellular Infiltration and Late-Phase Reaction
- 4–12 hours after exposure:
- Influx of eosinophils, neutrophils, and T-cells.
- Eosinophils release major basic protein (MBP) and eosinophil cationic protein (ECP) → damage epithelial cells.
- Leads to epithelial desquamation, sloughing, and increased airway sensitivity.
- Goblet cell hyperplasia → thick mucus plugs (often visible as Curschmann’s spirals).
- Charcot-Leyden crystals (eosinophil breakdown products) are seen in sputum.
4. Chronic Changes and Airway Remodeling
- Repeated inflammation causes permanent structural changes:
- Hypertrophy and hyperplasia of bronchial smooth muscle.
- Thickening of the basement membrane.
- Mucous gland hypertrophy.
- Increased angiogenesis (new blood vessel formation).
- Overall narrowing of airways and decreased reversibility.
- These changes may lead to chronic, persistent asthma and even fixed airflow obstruction, similar to COPD in late stages.
Summary in Chart Form
| Stage / Change | Key Features |
|---|---|
| Sensitization Phase | Th2 cell activation, IgE production, mast cell priming |
| Early Phase Reaction | Mast cell degranulation, bronchoconstriction, edema, mucus |
| Late Phase Reaction | Eosinophilic infiltration, epithelial damage, goblet cell hyperplasia |
| Airway Remodeling | Smooth muscle hypertrophy, basement membrane thickening, fibrosis |
Clinical and Nursing Relevance
- Wheezing, cough, chest tightness, and dyspnea are classic symptoms.
- Sputum may contain:
- Curschmann’s spirals, Charcot-Leyden crystals.
- Nurses play a key role in:
- Peak flow monitoring,
- Inhaler technique education,
- Trigger avoidance counseling,
- Monitoring for status asthmaticus (life-threatening asthma attack).
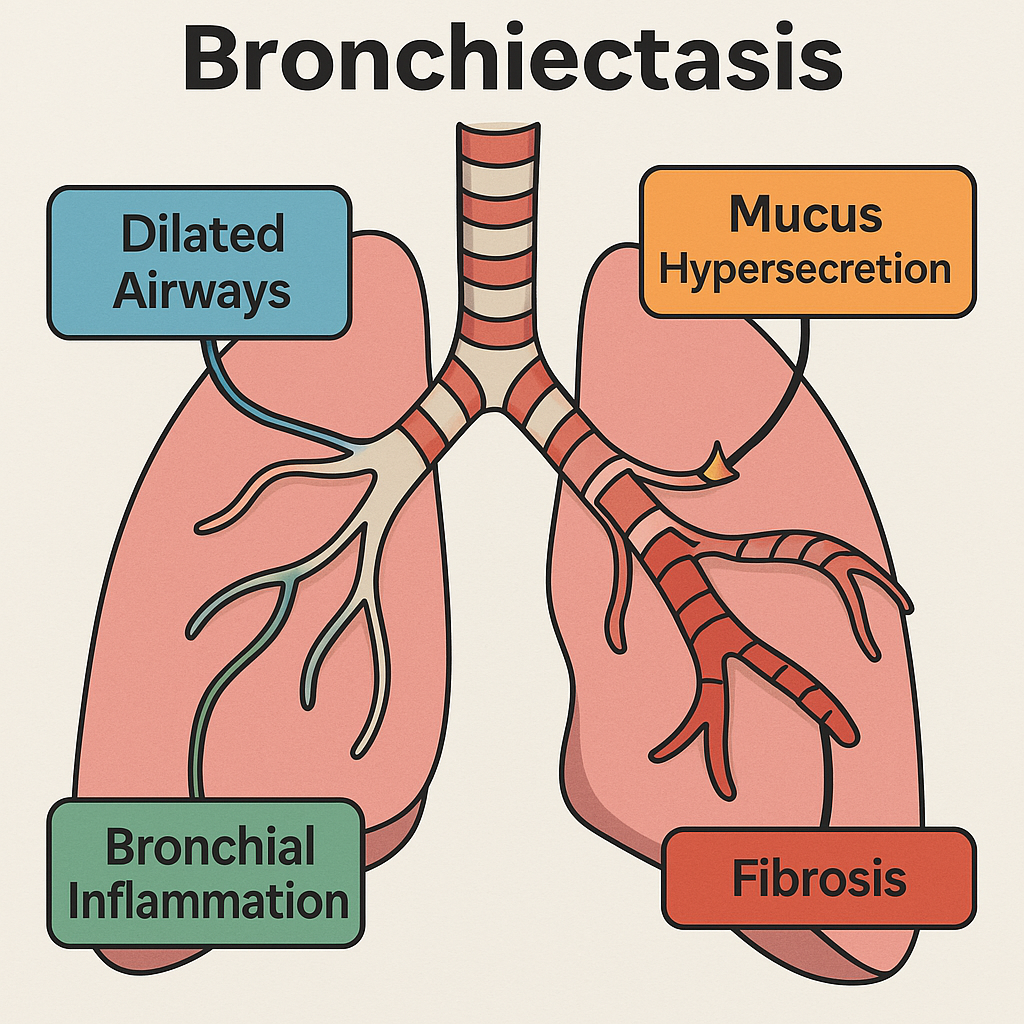
Pathological Changes in Bronchiectasis
Bronchiectasis is a chronic, irreversible condition involving permanent dilatation of bronchi and bronchioles due to destruction of the muscular and elastic components of their walls.
It is often the end-result of chronic or recurrent infections, particularly in settings of impaired mucociliary clearance, bronchial obstruction, or immune deficiency.
1. Initial Trigger: Infection, Obstruction, or Immune Deficiency
- Common Causes:
- Recurrent or severe respiratory infections (e.g., Mycobacterium tuberculosis, Staphylococcus, Haemophilus).
- Obstruction (foreign body, tumor, enlarged lymph nodes).
- Congenital conditions (e.g., cystic fibrosis, Kartagener’s syndrome).
- Immune compromise (e.g., hypogammaglobulinemia).
- Pathogenesis Begins with:
- Impaired clearance of mucus.
- Stagnation of secretions → bacterial colonization and persistent inflammation.
2. Inflammation and Wall Damage
- Persistent infection triggers:
- Chronic neutrophilic inflammation in the bronchial wall.
- Release of proteolytic enzymes (elastase, collagenase).
- These enzymes destroy the bronchial wall’s elastin, cartilage, and smooth muscle.
- This leads to:
- Structural weakness, loss of support,
- Bronchial wall collapse, and
- Progressive dilatation of airways.
3. Permanent Bronchial Dilatation and Mucus Plugging
- Damaged bronchi become distorted and permanently dilated.
- Classified morphologically as:
- Cylindrical (uniform dilation),
- Varicose (beaded, irregular walls),
- Saccular/Cystic (ballooned, grape-like appearance).
- Classified morphologically as:
- Mucus pools in the dilated areas, forming thick purulent plugs, which:
- Obstruct airflow,
- Serve as reservoirs for recurrent infection.
4. Fibrosis and Parenchymal Damage
- Repeated cycles of infection and inflammation extend into surrounding alveoli and interstitium.
- Leads to:
- Peribronchial fibrosis,
- Collapse of distal lung parenchyma (atelectasis),
- Ventilation-perfusion mismatch → hypoxia, clubbing.
- Over time, lung architecture becomes grossly distorted.
Summary in Chart Form
| Stage / Change | Key Pathological Features |
|---|---|
| Initiation | Infection, obstruction, or immune defect |
| Inflammatory Phase | Neutrophil-mediated wall destruction (enzymes degrade elastin/cartilage) |
| Bronchial Dilatation | Cylindrical, varicose, or cystic dilation with mucus pooling |
| Fibrosis and Lung Destruction | Peribronchial fibrosis, atelectasis, recurrent infection |
Clinical and Nursing Relevance
- Symptoms: Chronic productive cough, large volume of purulent sputum, hemoptysis, wheezing, clubbing.
- Radiological findings: Tram-track appearance, signet-ring sign on HRCT chest.
- Nursing care:
- Promote airway clearance (postural drainage, chest physiotherapy),
- Antibiotic adherence,
- Monitor for oxygen therapy, nutrition, and infection prevention.
Pathological Changes in Tumors of the Lungs
Lung tumors are abnormal proliferations of cells in the pulmonary tissues. They can be benign (non-invasive, localized) or malignant (invasive, potentially metastatic). Lung cancer, particularly the malignant type, is one of the most common and deadliest cancers worldwide.
Most primary malignant lung tumors are broadly classified into:
- Non-Small Cell Lung Carcinoma (NSCLC) – ~85%
- Small Cell Lung Carcinoma (SCLC) – ~15%
1. Initiation: Genetic Mutations and Carcinogen Exposure
- Causes:
- Cigarette smoking (main risk factor),
- Exposure to asbestos, radon gas, air pollutants, industrial carcinogens,
- Genetic predisposition (EGFR, KRAS, ALK mutations).
- Pathological Events:
- DNA damage in epithelial cells → oncogene activation and tumor suppressor gene inactivation (e.g., p53).
- Uncontrolled cell proliferation, apoptosis inhibition, and loss of differentiation.
2. Early Tumor Formation and Local Growth
- Origin:
- Begins in bronchial epithelium, alveolar cells, or submucosal glands.
- Formation of a neoplastic mass or polypoid lesion projecting into the bronchial lumen or infiltrating parenchyma.
- Local Changes:
- Destruction of bronchial wall, narrowing or obstruction of airways.
- Associated atelectasis, post-obstructive pneumonia, or bronchiectasis.
- May invade pleura, chest wall, or blood vessels.
- Histological Types:
- Squamous cell carcinoma: Keratin pearls, intercellular bridges.
- Adenocarcinoma: Gland-forming, mucin-producing cells.
- Small cell carcinoma: Dense small cells with scant cytoplasm, high mitosis.
- Large cell carcinoma: Poorly differentiated, lacks features of other types.
3. Invasion and Angiogenesis
- As tumor enlarges, it gains the ability to:
- Invade surrounding structures (pleura, pericardium, lymphatics).
- Stimulate angiogenesis via VEGF (vascular endothelial growth factor) → increased blood supply to support growth.
- Tumor necrosis may occur due to rapid growth outstripping blood supply.
4. Metastasis (Regional and Distant)
- Lymphatic Spread:
- Commonly to hilar and mediastinal lymph nodes.
- Hematogenous Spread:
- Frequent sites: brain, liver, bones, adrenal glands.
- Pleural Involvement:
- Tumors may seed the pleural space → malignant pleural effusion.
5. Paraneoplastic Syndromes and Systemic Effects
- Tumors can secrete hormones or hormone-like substances → paraneoplastic syndromes:
- SIADH, Cushing’s syndrome, hypercalcemia, clubbing, neurologic syndromes.
- Cachexia, anemia, and fatigue occur as systemic manifestations.
Summary in Chart Form
| Stage / Feature | Key Pathological Changes |
|---|---|
| Initiation | DNA mutations from smoking/carcinogens; oncogene activation |
| Local Tumor Formation | Bronchial epithelial mass, airway obstruction, local invasion |
| Invasion and Angiogenesis | Destruction of tissue, new blood vessels, tumor necrosis |
| Metastasis | Spread to lymph nodes, brain, bone, liver, adrenals |
| Paraneoplastic/Systemic Effects | Hormone secretion, weight loss, fatigue, clubbing |
Clinical and Nursing Relevance
- Symptoms: Persistent cough, hemoptysis, weight loss, hoarseness, dyspnea.
- Diagnosis: Chest X-ray, CT scan, bronchoscopy with biopsy, PET scan.
- Nursing Roles:
- Early symptom identification,
- Patient education and emotional support,
- Monitoring response to chemo/radiation therapy,
- Managing side effects and palliative care.
Pathological Changes in Tumors of the Lungs
Lung tumors are abnormal proliferations of cells in the pulmonary tissues. They can be benign (non-invasive, localized) or malignant (invasive, potentially metastatic). Lung cancer, particularly the malignant type, is one of the most common and deadliest cancers worldwide.
Most primary malignant lung tumors are broadly classified into:
- Non-Small Cell Lung Carcinoma (NSCLC) – ~85%
- Small Cell Lung Carcinoma (SCLC) – ~15%
1. Initiation: Genetic Mutations and Carcinogen Exposure
- Causes:
- Cigarette smoking (main risk factor),
- Exposure to asbestos, radon gas, air pollutants, industrial carcinogens,
- Genetic predisposition (EGFR, KRAS, ALK mutations).
- Pathological Events:
- DNA damage in epithelial cells → oncogene activation and tumor suppressor gene inactivation (e.g., p53).
- Uncontrolled cell proliferation, apoptosis inhibition, and loss of differentiation.
2. Early Tumor Formation and Local Growth
- Origin:
- Begins in bronchial epithelium, alveolar cells, or submucosal glands.
- Formation of a neoplastic mass or polypoid lesion projecting into the bronchial lumen or infiltrating parenchyma.
- Local Changes:
- Destruction of bronchial wall, narrowing or obstruction of airways.
- Associated atelectasis, post-obstructive pneumonia, or bronchiectasis.
- May invade pleura, chest wall, or blood vessels.
- Histological Types:
- Squamous cell carcinoma: Keratin pearls, intercellular bridges.
- Adenocarcinoma: Gland-forming, mucin-producing cells.
- Small cell carcinoma: Dense small cells with scant cytoplasm, high mitosis.
- Large cell carcinoma: Poorly differentiated, lacks features of other types.
3. Invasion and Angiogenesis
- As tumor enlarges, it gains the ability to:
- Invade surrounding structures (pleura, pericardium, lymphatics).
- Stimulate angiogenesis via VEGF (vascular endothelial growth factor) → increased blood supply to support growth.
- Tumor necrosis may occur due to rapid growth outstripping blood supply.
4. Metastasis (Regional and Distant)
- Lymphatic Spread:
- Commonly to hilar and mediastinal lymph nodes.
- Hematogenous Spread:
- Frequent sites: brain, liver, bones, adrenal glands.
- Pleural Involvement:
- Tumors may seed the pleural space → malignant pleural effusion.
5. Paraneoplastic Syndromes and Systemic Effects
- Tumors can secrete hormones or hormone-like substances → paraneoplastic syndromes:
- SIADH, Cushing’s syndrome, hypercalcemia, clubbing, neurologic syndromes.
- Cachexia, anemia, and fatigue occur as systemic manifestations.
Summary in Chart Form
| Stage / Feature | Key Pathological Changes |
|---|---|
| Initiation | DNA mutations from smoking/carcinogens; oncogene activation |
| Local Tumor Formation | Bronchial epithelial mass, airway obstruction, local invasion |
| Invasion and Angiogenesis | Destruction of tissue, new blood vessels, tumor necrosis |
| Metastasis | Spread to lymph nodes, brain, bone, liver, adrenals |
| Paraneoplastic/Systemic Effects | Hormone secretion, weight loss, fatigue, clubbing |
Clinical and Nursing Relevance
- Symptoms: Persistent cough, hemoptysis, weight loss, hoarseness, dyspnea.
- Diagnosis: Chest X-ray, CT scan, bronchoscopy with biopsy, PET scan.
- Nursing Roles:
- Early symptom identification,
- Patient education and emotional support,
- Monitoring response to chemo/radiation therapy,
- Managing side effects and palliative care.

Cardio-vascular system
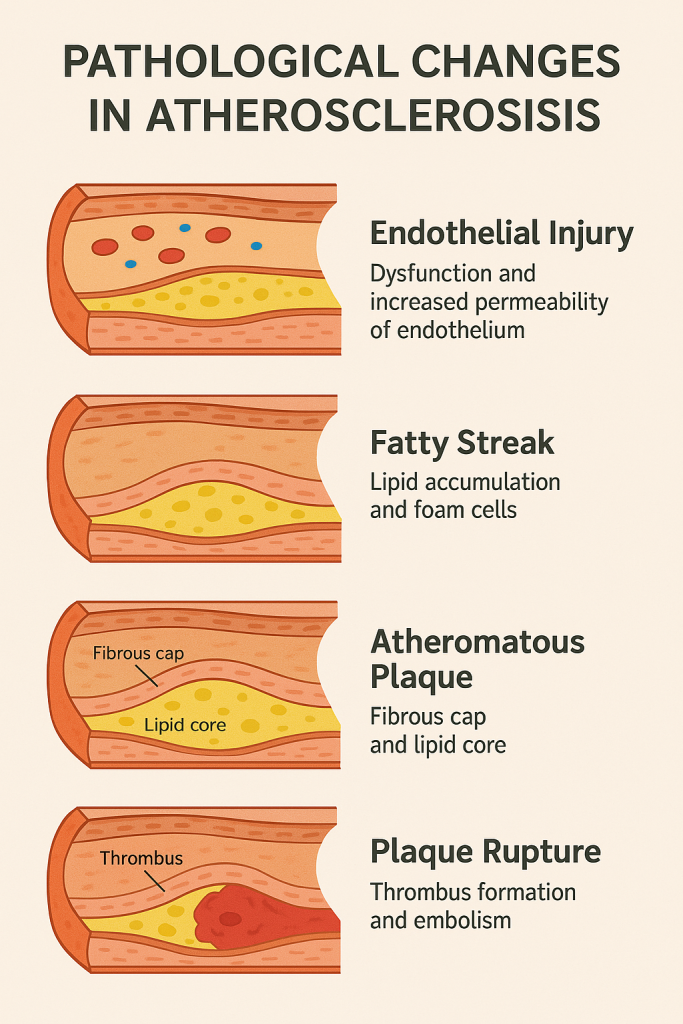
Pathological Changes in Atherosclerosis
Atherosclerosis is a chronic progressive disease of large and medium-sized arteries characterized by formation of fibrofatty plaques (atheromas) in the intimal layer, leading to narrowing of the arterial lumen, reduced elasticity, and compromised blood flow.
It is the underlying cause of major cardiovascular diseases such as coronary artery disease, cerebrovascular disease, and peripheral artery disease.
1. Endothelial Injury – The Initiating Event
- Triggers:
- Hypertension,
- Hyperlipidemia (especially LDL cholesterol),
- Smoking,
- Diabetes,
- Infections or toxins.
- Pathology:
- Endothelial cells become dysfunctional and lose their ability to:
- Maintain vascular tone,
- Prevent platelet adhesion,
- Inhibit smooth muscle proliferation.
- Endothelial cells become dysfunctional and lose their ability to:
- Result:
- Increased vascular permeability, leukocyte adhesion, and migration of monocytes into the intima.
2. Fatty Streak Formation (Lipid Accumulation and Foam Cells)
- LDL cholesterol enters the intima and gets oxidized.
- Attracted monocytes differentiate into macrophages, which engulf oxidized LDL → become foam cells.
- Clusters of foam cells form fatty streaks, the earliest visible lesion of atherosclerosis.
- Reversible Stage: Fatty streaks can regress with lifestyle and lipid control.
3. Formation of Atheromatous Plaque (Fibrous Cap and Lipid Core)
- Continued inflammation triggers:
- Smooth muscle cell migration from media to intima.
- Collagen and proteoglycan production, forming a fibrous cap.
- Accumulation of foam cells, lipids, and debris forms a necrotic lipid core.
- Plaque Structure:
- Fibrous cap (collagen + smooth muscle)
- Lipid-rich necrotic core
- Shoulder region (rich in macrophages, T cells)
- Effect: Arterial lumen narrows → ischemia, especially during increased demand.
4. Plaque Complications and Rupture
- Plaques can become unstable due to:
- Thin fibrous cap,
- Active inflammation,
- Enzymatic degradation.
- Consequences:
- Plaque rupture exposes thrombogenic material.
- Platelet adhesion → thrombus formation → acute infarction.
- Aneurysm formation due to weakening of vessel wall.
Summary in Chart Form
| Stage / Change | Key Pathological Features |
|---|---|
| Endothelial Injury | Dysfunction, ↑ permeability, leukocyte adhesion |
| Fatty Streaks | Foam cell accumulation, early lipid deposits |
| Atheromatous Plaque | Fibrous cap + necrotic lipid core, smooth muscle involvement |
| Plaque Rupture / Thrombosis | Thrombus formation, embolism, infarction, aneurysm risk |
Clinical and Nursing Relevance
- Common sites: Coronary arteries, carotid arteries, abdominal aorta.
- Complications: Myocardial infarction, stroke, gangrene, aneurysms.
- Nursing care:
- Lifestyle education: diet, exercise, smoking cessation.
- Medication monitoring (statins, antiplatelets).
- Patient counseling on warning signs of MI/stroke.
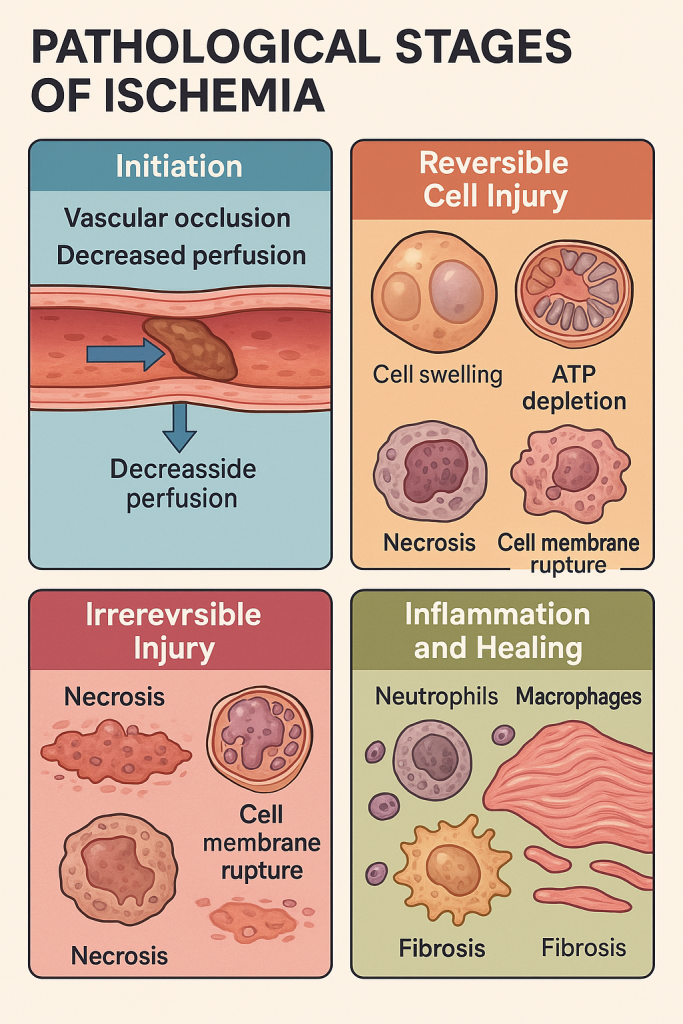
Pathological Changes in Ischemia
Ischemia is defined as a localized reduction or complete interruption of blood flow to a tissue or organ, leading to oxygen and nutrient deprivation. Unlike hypoxia, which is a general lack of oxygen, ischemia involves both lack of oxygen (O₂) and accumulation of metabolic waste, due to compromised perfusion.
It is a reversible process initially, but prolonged ischemia can lead to irreversible cell injury, necrosis, and organ dysfunction.
1. Vascular Occlusion or Perfusion Impairment – Initiation of Ischemia
- Causes:
- Thrombus or embolus
- Atherosclerosis
- Vasospasm
- Compression (e.g., from tumors, edema)
- Shock or systemic hypotension
- Pathophysiology:
- Blood flow is partially or completely blocked.
- Affected tissue suffers acute shortage of oxygen (hypoxia) and nutrients like glucose.
- Anaerobic metabolism is activated → lactic acid buildup → cellular acidosis.
2. Reversible Cellular Injury (Early Phase)
- Duration: Within 15–30 minutes of ischemia (variable by tissue type).
- Cellular Changes:
- ↓ ATP → Failure of Na⁺/K⁺ pumps → cellular swelling (hydropic change)
- Mitochondrial swelling, ribosomal detachment → ↓ protein synthesis
- Chromatin clumping, loss of membrane integrity
- Functional Impairment: Organ dysfunction begins (e.g., angina, TIA)
3. Irreversible Injury and Necrosis (Prolonged Ischemia)
- Duration: Varies by tissue:
- Brain: 3–5 minutes
- Myocardium: ~20–30 minutes
- Kidney and liver: 1–2 hours
- Pathological Features:
- Cell membrane rupture → leakage of contents
- Lysosomal enzyme release → autolysis
- Inflammatory cell infiltration
- Coagulative necrosis in most tissues
- Liquefactive necrosis in brain tissue
- Gross Appearance:
- Pale, firm tissue (white infarct in heart, kidney)
- Red infarct in dual-blood-supply organs (lungs, intestines)
4. Inflammation and Healing Response
- Neutrophil infiltration within 12–24 hours
- Followed by macrophage clearance, granulation tissue formation
- If tissue survives: Regeneration or Fibrosis/Scarring
Summary in Chart Form
| Stage / Change | Key Pathological Features |
|---|---|
| Initiation | Vascular blockage, ↓ perfusion, hypoxia |
| Reversible Injury | ATP depletion, cell swelling, mitochondrial dysfunction |
| Irreversible Injury & Necrosis | Membrane rupture, enzyme leakage, coagulative necrosis, inflammation |
| Inflammation and Healing | Neutrophils → macrophages → granulation tissue → fibrosis or regeneration |
Clinical and Nursing Relevance
- Common ischemic conditions:
- Myocardial infarction (heart)
- Cerebral infarction (stroke)
- Bowel ischemia, renal infarction, limb gangrene
- Nursing care includes:
- Early symptom recognition (pain, pallor, coldness, neurologic signs)
- Monitoring vital signs, oxygenation
- Administering antiplatelets, thrombolytics, and preparing for revascularization procedures
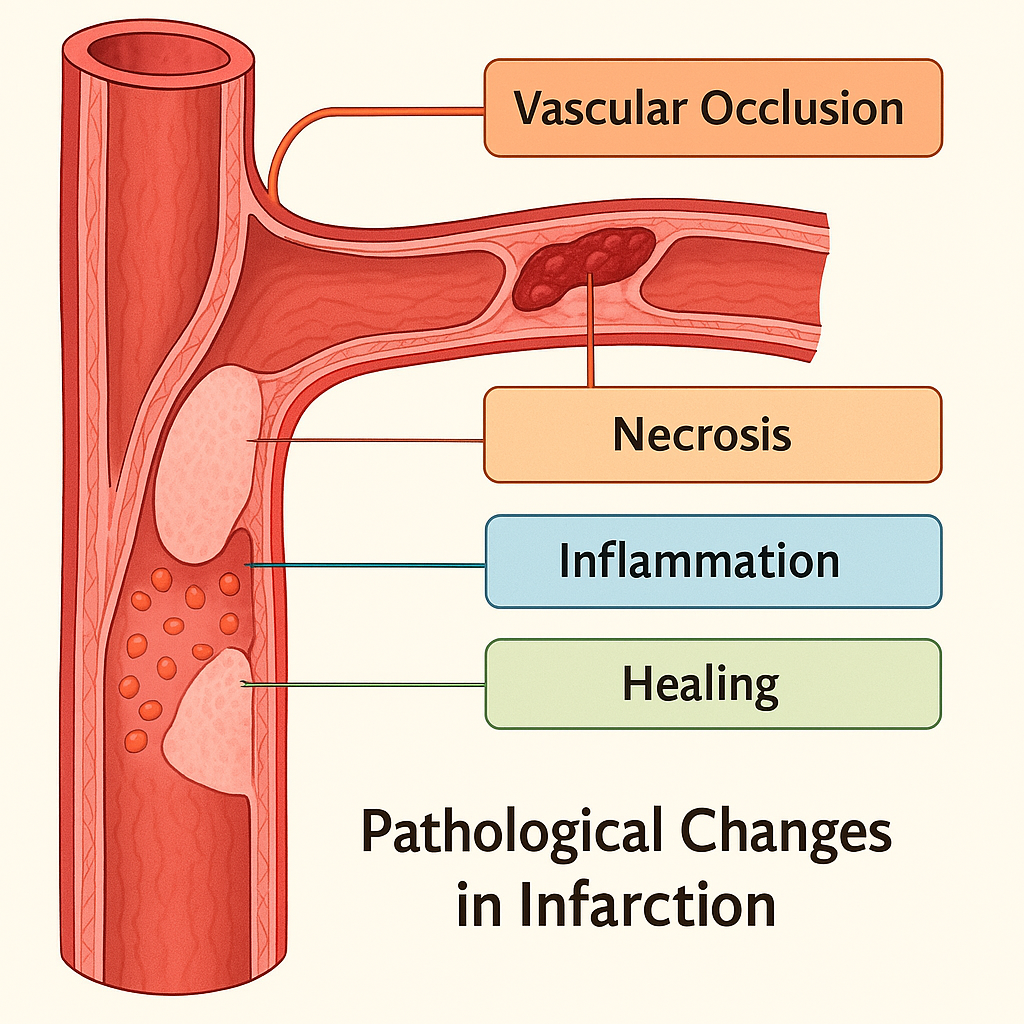
Pathological Changes in Infarction
Infarction refers to tissue necrosis caused by prolonged ischemia, due to obstruction of the arterial blood supply or venous drainage. It is an irreversible consequence of sustained ischemia and is one of the leading causes of death globally, especially in myocardial and cerebral infarctions.
1. Vascular Occlusion: Initiation of Infarction
- Causes:
- Arterial thrombus or embolus
- Atherosclerotic plaque rupture
- Venous thrombosis (less common, often in organs with a single venous outflow)
- External compression (e.g., volvulus, hernia)
- Pathological Process:
- Blocked vessel → decreased perfusion → ischemia
- If unrelieved: oxygen and nutrient deprivation → cell death
2. Progressive Tissue Hypoxia and Necrosis
- Ischemic necrosis begins within:
- Brain: 3–5 minutes
- Heart: ~20–30 minutes
- Kidneys/Spleen: 1–2 hours
- Necrosis type:
- Coagulative necrosis (most tissues)
- Liquefactive necrosis (brain)
- Hemorrhagic necrosis (in organs with dual blood supply, e.g., lungs)
- Gross appearance varies:
- Pale infarct: seen in solid organs like heart, kidney, spleen.
- Red infarct: seen in lungs, intestine (due to dual blood supply or venous congestion).
3. Inflammation and Cellular Response
- Within 6–12 hours:
- Neutrophils infiltrate the necrotic area.
- Followed by macrophages which phagocytose dead cells.
- Inflammatory response also involves cytokine release, attracting further immune cells.
- Clinical relevance: Can cause fever, pain, and leukocytosis.
4. Healing, Scarring, or Complications
- Small infarcts may be resorbed.
- Large infarcts → replaced by fibrous scar via granulation tissue.
- Complications:
- Rupture (e.g., myocardial free wall rupture)
- Cystic cavity formation (in brain)
- Infection and abscess formation
- Contracture or organ dysfunction (chronic infarcts)
Summary in Chart Form
| Stage / Change | Key Pathological Features |
|---|---|
| Vascular Occlusion | Thrombus, embolus, or compression leads to ischemia |
| Necrosis | Coagulative (heart), liquefactive (brain), hemorrhagic (lungs) |
| Inflammatory Response | Neutrophils → macrophages, cytokine-mediated reaction |
| Healing & Scarring | Fibrosis, complications like rupture or abscess |
Clinical and Nursing Relevance
- Common infarcts: Myocardial infarction, stroke, pulmonary infarction, renal and splenic infarcts.
- Symptoms depend on organ: Chest pain (heart), neurological deficits (brain), hemoptysis (lung).
- Nursing care:
- Early recognition of signs/symptoms
- Monitor vitals, ECG/CT/MRI reports
- Supportive care, oxygenation, thrombolytics/anticoagulants
- Educate on lifestyle modification and medication adherence
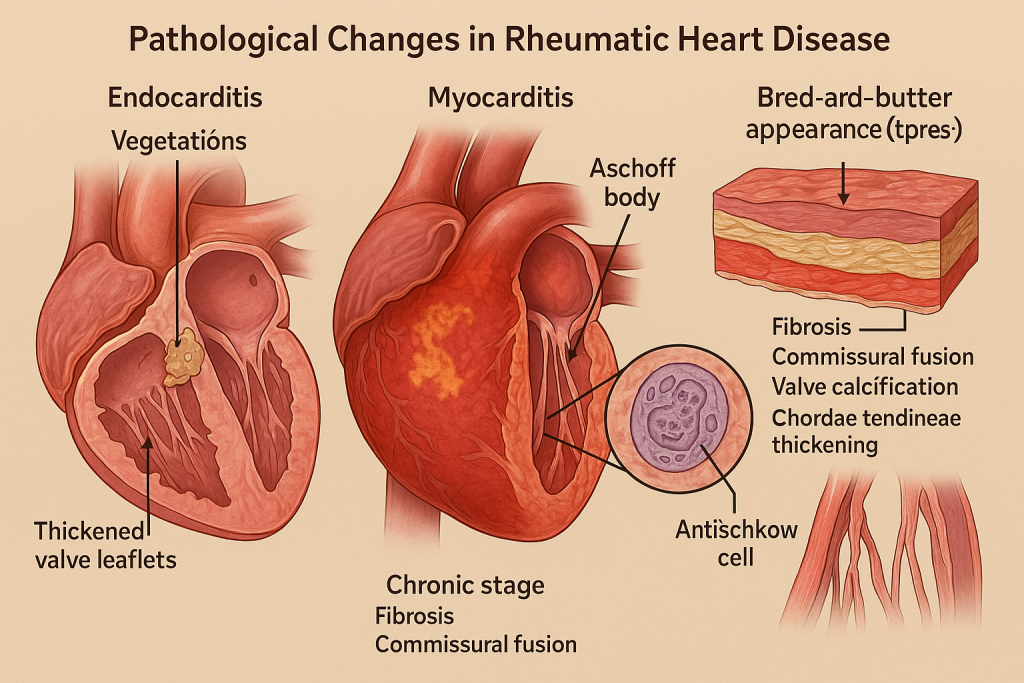
Pathological Changes in Rheumatic Heart Disease
Rheumatic Heart Disease (RHD) is a chronic valvular complication that develops after Acute Rheumatic Fever (ARF), an autoimmune response to group A β-hemolytic streptococcal infection (typically pharyngitis). It primarily affects children and adolescents in low-resource settings and is a major cause of cardiovascular morbidity worldwide.
The disease is characterized by immune-mediated inflammation of cardiac tissues—endocardium, myocardium, and pericardium—and progressive scarring of heart valves, especially the mitral valve.
1. Acute Phase: Pancarditis (All Three Layers Affected)
① Endocarditis (valves)
- Earliest lesion involves fibrinoid necrosis along the lines of valve closure.
- Vegetations (verrucae) form on mitral and aortic valves.
- Commonly affected valves:
- Mitral (most common) → Mitral stenosis
- Aortic, Tricuspid, Pulmonary (rare)
② Myocarditis
- Formation of Aschoff bodies:
- Pathognomonic lesions composed of central fibrinoid necrosis, Anitschkow cells (activated macrophages), lymphocytes, and plasma cells.
- Causes flabby myocardium → may lead to conduction defects or heart failure.
③ Pericarditis
- Leads to fibrinous or serofibrinous pericarditis.
- “Bread-and-butter pericarditis” appearance grossly.
- Usually self-limiting.
2. Chronic Phase: Progressive Valve Damage and Fibrosis
- Healing process results in:
- Fibrosis and thickening of valve leaflets,
- Fusion of commissures,
- Shortening and fusion of chordae tendineae,
- Calcification of valves.
- Valvular dysfunctions:
- Mitral stenosis (narrowed orifice, “fish mouth” appearance)
- Mitral regurgitation
- Aortic valve involvement → Aortic stenosis or regurgitation
- May lead to:
- Left atrial enlargement, pulmonary hypertension, right heart failure, atrial fibrillation, and thromboembolism.
3. Histopathological Findings
- Aschoff bodies in myocardium (central necrosis + Anitschkow cells).
- Verrucae on valve leaflets (tiny, warty vegetations).
- Fibrous thickening, hyalinization, and calcification of valves.
- Infiltration with T-cells, plasma cells, macrophages.
Summary in Chart Form
| Stage / Structure Affected | Key Pathological Features |
|---|---|
| Endocardium | Vegetations, valve thickening, commissural fusion (especially mitral) |
| Myocardium | Aschoff bodies, Anitschkow cells, myocarditis → arrhythmia or failure |
| Pericardium | Fibrinous pericarditis (“bread and butter” appearance) |
| Chronic Stage | Valve scarring, stenosis/regurgitation, atrial enlargement, fibrosis |
Clinical and Nursing Relevance
- Symptoms: Dyspnea, palpitations, chest discomfort, murmur (diastolic or systolic).
- Signs: Malar flush (mitral facies), loud S1, opening snap (mitral stenosis).
- Nursing Care:
- Ensure prophylactic antibiotics to prevent recurrence of streptococcal infection.
- Monitor cardiac function, signs of heart failure.
- Educate patient on medication adherence, endocarditis prophylaxis, lifestyle adjustments.
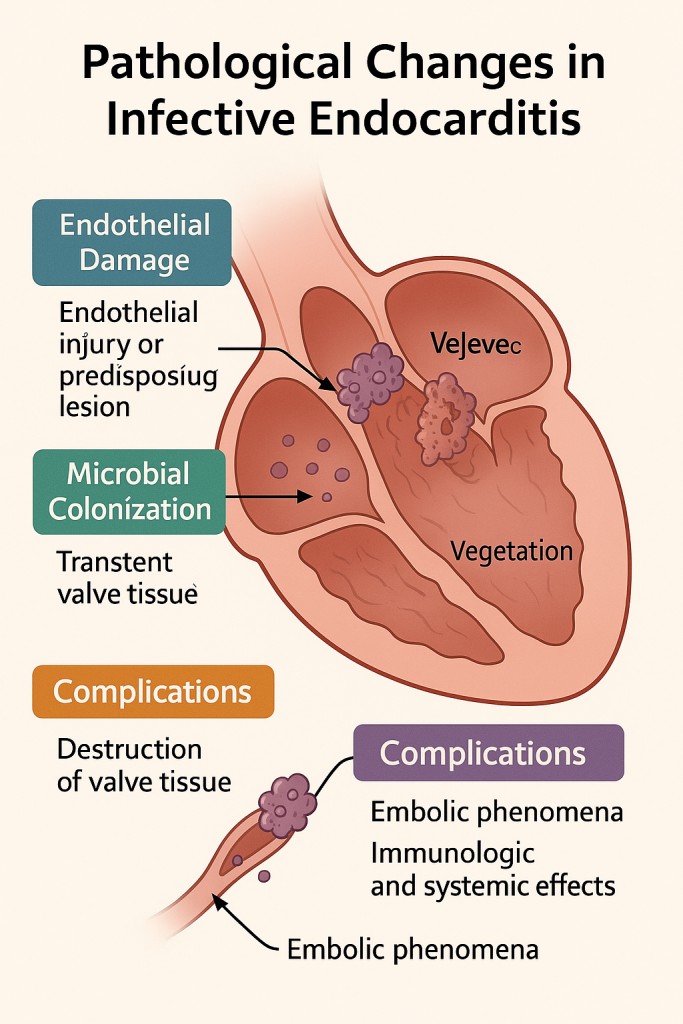
Pathological Changes in Infective Endocarditis
Infective endocarditis is a microbial infection of the endocardial surface of the heart, most commonly affecting the heart valves. It is characterized by vegetations composed of thrombotic debris and organisms, and can cause destruction of cardiac tissue and systemic embolization.
It may be acute (rapid, aggressive, often in normal valves) or subacute/chronic (slow, often on damaged valves).
1. Endothelial Damage or Predisposing Lesion – Initiating Event
- Risk Factors:
- Pre-existing valvular abnormalities (e.g., RHD, MVP)
- Prosthetic heart valves
- Congenital heart defects (e.g., VSD)
- IV drug use, invasive procedures
- Mechanism:
- Endothelial injury → exposure of subendothelial collagen and tissue factor → platelet and fibrin deposition.
- Forms sterile thrombotic vegetations (Non-bacterial thrombotic endocarditis – NBTE).
2. Microbial Colonization and Vegetation Formation
- Transient bacteremia (e.g., after dental or surgical procedures) seeds the sterile vegetations.
- Microorganisms (e.g., Staphylococcus aureus, Streptococcus viridans) adhere and multiply.
- Vegetations form:
- Composed of fibrin, inflammatory cells, platelets, and colonies of organisms.
- Typically located on the line of closure of affected valve cusps.
- Friable, bulky, and destructive in acute cases (can cause valve rupture).
3. Valve and Cardiac Tissue Destruction
- In acute IE:
- Vegetations rapidly destroy valve cusps → acute regurgitation, heart failure.
- Extension into adjacent myocardium can lead to ring abscesses, perforations, or fistulas.
- In subacute IE:
- Slower tissue damage,
- Formation of granulation tissue, fibrosis, and calcification.
- Most affected valves:
- Mitral > Aortic > Tricuspid (especially in IV drug users)
4. Complications: Embolism, Immunologic, and Systemic
- Embolic phenomena:
- Vegetations can break off → emboli to brain (stroke), lungs, spleen, kidneys, extremities.
- Infarcts or abscesses in affected organs.
- Immunologic complications:
- Circulating immune complexes → glomerulonephritis, vasculitis, Osler nodes, Roth spots.
- Sepsis or metastatic infections may develop (e.g., vertebral osteomyelitis, septic arthritis).
Summary in Chart Form
| Stage / Pathological Change | Key Features |
|---|---|
| Endothelial Damage | Injury → NBTE → platelet/fibrin deposition |
| Microbial Colonization | Bacteremia seeds thrombi → vegetations with organisms |
| Valve Destruction | Tissue necrosis, perforation, ring abscess, regurgitation |
| Complications | Embolism, immune complex disease, infarcts, sepsis |
Clinical and Nursing Relevance
- Symptoms: Fever, murmur, petechiae, splinter hemorrhages, clubbing.
- Signs: Osler nodes (painful), Janeway lesions (painless), Roth spots, splenomegaly.
- Diagnosis: Blood cultures, echocardiogram (TTE/TEE), Duke criteria.
- Nursing Care:
- Prompt collection of cultures before antibiotics.
- Monitor for signs of embolism or heart failure.
- Educate on antibiotic prophylaxis for high-risk patients.
Gastrointestinal tract
Pathological Changes in Peptic Ulcer
A peptic ulcer is a localized breach in the mucosa of the stomach or duodenum caused by the acid-pepsin digestive action on the epithelium. It is a chronic condition resulting from an imbalance between mucosal defensive factors and aggressive factors like acid, pepsin, Helicobacter pylori, and NSAIDs.
Peptic ulcers most commonly occur in:
- The duodenum (first part)
- The lesser curvature of the stomach
1. Mucosal Injury and Imbalance of Defense vs. Aggression
- Defensive Factors:
- Mucus-bicarbonate barrier,
- Surface epithelial integrity,
- Prostaglandins,
- Adequate blood flow.
- Aggressive Factors:
- Gastric acid and pepsin,
- H. pylori infection: disrupts mucosal barrier and induces inflammation,
- NSAIDs: inhibit prostaglandins, reducing mucosal protection,
- Smoking, stress, alcohol.
- The result is a focal loss of mucosal integrity leading to erosion and ulcer formation.
2. Ulcer Formation and Layered Destruction
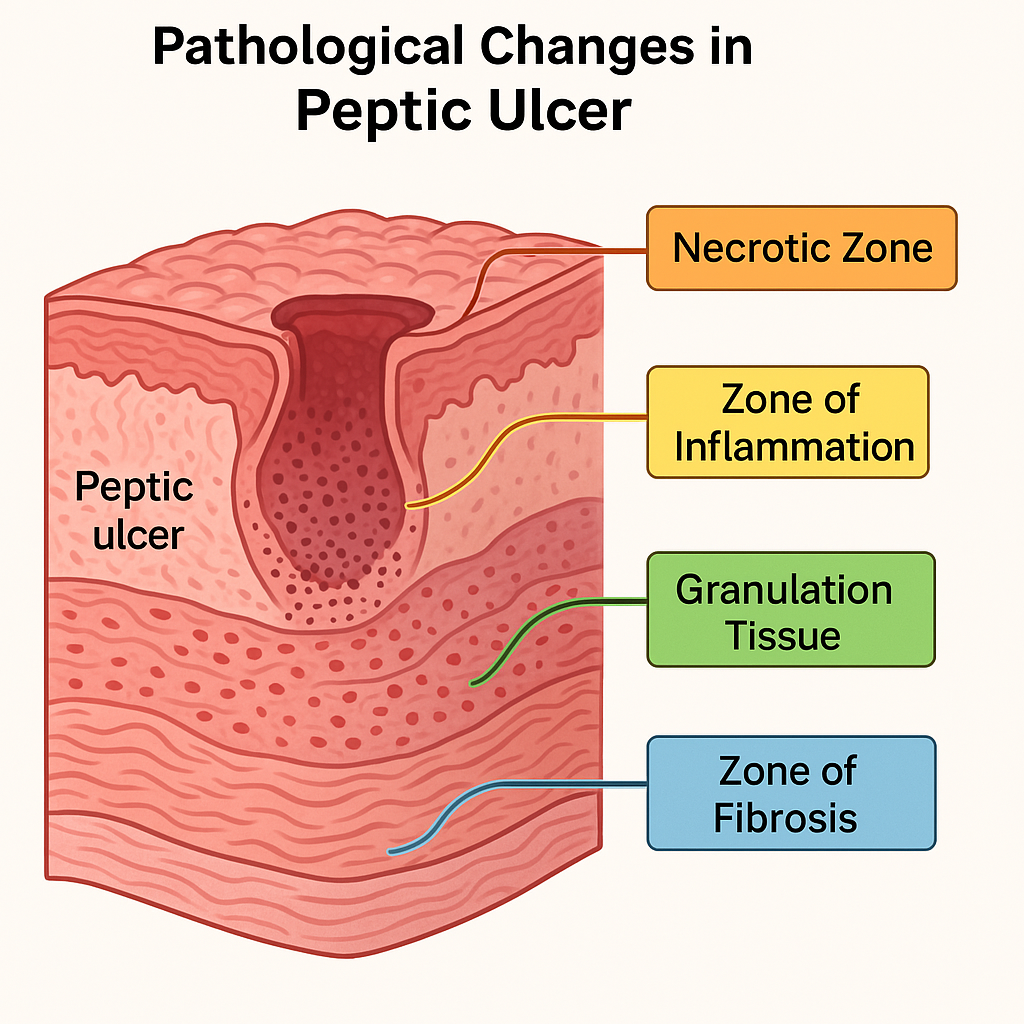
Peptic ulcers penetrate the mucosa and can extend into deeper layers of the GI wall. The histological architecture of a peptic ulcer typically has four distinct layers from superficial to deep:
Layer 1: Necrotic Zone
- Thin superficial zone with coagulative necrosis.
- Composed of dead epithelial cells, debris, and denatured proteins.
Layer 2: Zone of Inflammation
- Dense infiltration of neutrophils, lymphocytes, and macrophages.
- Inflammatory cells attempt to contain tissue damage.
Layer 3: Granulation Tissue
- Proliferation of fibroblasts, capillaries, and chronic inflammatory cells.
- Represents the body’s attempt to heal.
Layer 4: Zone of Fibrosis (Scar Tissue)
- Collagen deposition and fibrosis.
- Leads to rigidity of the wall and scar contraction in chronic ulcers.
3. Complications of Peptic Ulcer
- Hemorrhage: Erosion of a blood vessel leads to hematemesis or melena.
- Perforation: Full-thickness penetration → peritonitis.
- Penetration: Ulcer invades adjacent organs (e.g., pancreas, liver).
- Obstruction: Fibrotic scarring leads to gastric outlet obstruction.
Summary in Chart Form
| Stage / Layer | Pathological Features |
|---|---|
| Necrotic Zone | Superficial dead tissue, coagulative necrosis |
| Inflammation Zone | Neutrophilic and lymphocytic infiltrate |
| Granulation Tissue | Capillaries, fibroblasts, healing attempt |
| Fibrosis | Collagen scar, wall thickening, deformity |
| Complications | Bleeding, perforation, obstruction, penetration |
Clinical and Nursing Relevance
- Symptoms: Epigastric pain (related to meals), nausea, bloating, weight loss.
- Signs of complications: Tarry stools, sudden severe abdominal pain, vomiting.
- Diagnosis: Endoscopy with biopsy, urea breath test for H. pylori.
- Nursing Care:
- Monitor for bleeding signs.
- Educate on avoiding NSAIDs, alcohol, smoking.
- Ensure H. pylori eradication therapy adherence.
- Promote regular meals and stress reduction.
Pathological Changes in Gastric and Duodenal Ulcers
Gastric and duodenal ulcers are types of peptic ulcers, which are focal breaches in the gastrointestinal mucosa due to digestive action of acid and pepsin. While they share a common pathogenesis, their location, risk factors, clinical features, and complications vary slightly.
1. Etiological Basis and Common Pathogenesis
- Aggressive Factors:
- Helicobacter pylori infection (most common),
- Hyperacidity (especially in duodenal ulcers),
- NSAIDs (aspirin, ibuprofen),
- Smoking, alcohol, stress.
- Defensive Impairments:
- Impaired mucus-bicarbonate barrier,
- Reduced prostaglandin synthesis,
- Poor epithelial regeneration,
- Ischemia or poor mucosal perfusion.
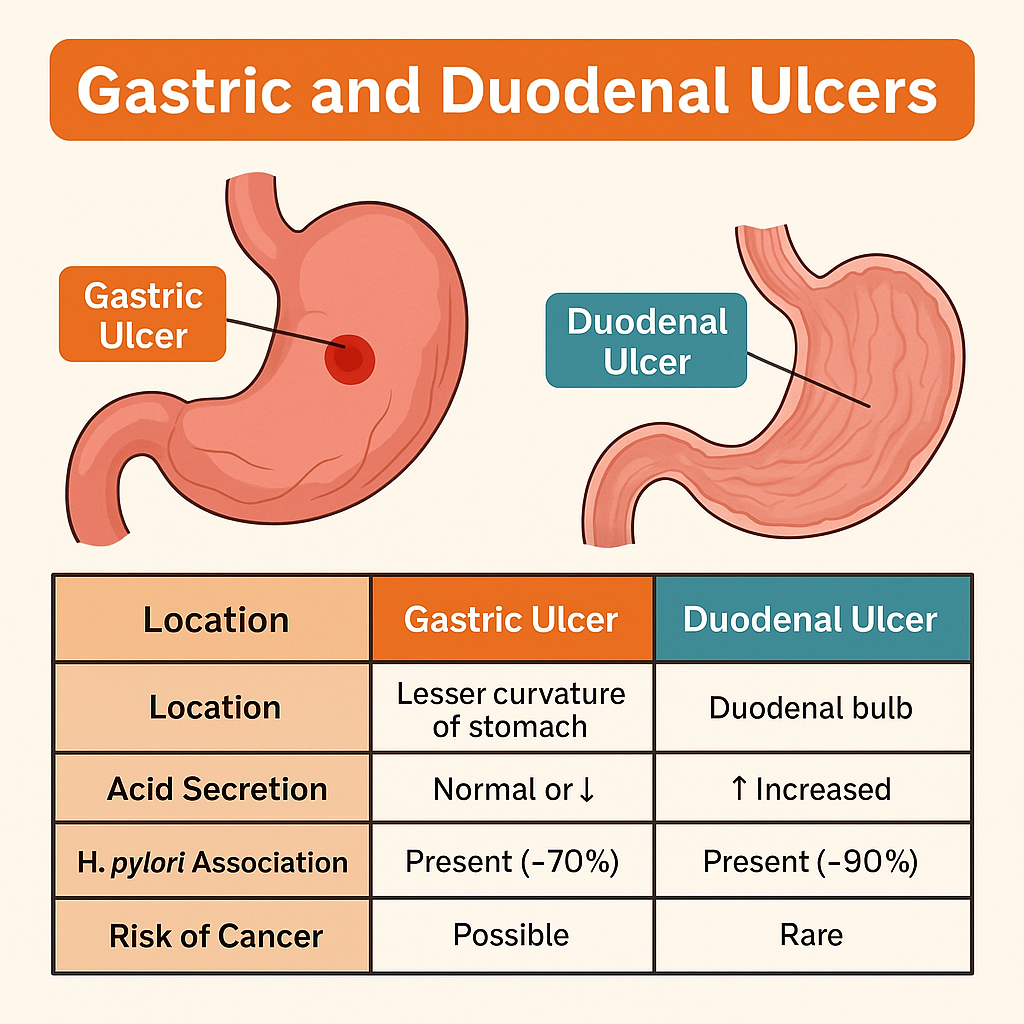
2. Location and Ulcer Characteristics
Gastric Ulcer
- Location: Usually along the lesser curvature of the stomach.
- Often occurs in older adults, especially those using NSAIDs.
- Associated with normal or decreased acid secretion.
Duodenal Ulcer
- Location: First part of the duodenum (bulb).
- Affects younger individuals, more acid-driven.
- Increased gastric acid output, especially at night.
- Stronger association with H. pylori.
3. Pathological Layers of Ulcer (Shared Histology)
Peptic ulcers (both gastric and duodenal) show four classic layers:
Necrotic Zone
- Surface layer composed of dead cells and denatured proteins due to acid-pepsin action.
Zone of Inflammation
- Dense infiltrate of neutrophils, lymphocytes, and plasma cells.
- Surrounds the necrotic debris.
Granulation Tissue Layer
- Proliferating capillaries, fibroblasts, and chronic inflammatory cells.
- Attempts to heal the ulcer.
Zone of Fibrosis
- Scar tissue replaces damaged tissue.
- May cause deformity or pyloric stenosis in chronic ulcers.
4. Complications
- Hemorrhage: Common in both; may cause hematemesis (gastric) or melena (duodenal).
- Perforation: Especially duodenal; leads to peritonitis.
- Obstruction: Due to fibrosis and inflammation at the pylorus.
- Malignancy:
- Gastric ulcers have a risk of malignant transformation.
- Duodenal ulcers are rarely malignant.
Summary in Chart Form
| Feature | Gastric Ulcer | Duodenal Ulcer |
|---|---|---|
| Location | Lesser curvature of stomach | Duodenal bulb |
| Acid Secretion | Normal or ↓ | ↑ Increased |
| Age Group | Older adults | Younger individuals |
| H. pylori Association | Present (~70%) | Present (~90%) |
| Risk of Cancer | Possible (must biopsy) | Rare |
| Pain Timing | Worsens with food | Improves with food, worse at night |
| Common Complications | Bleeding, perforation, carcinoma | Bleeding, perforation, fibrosis |
Clinical and Nursing Relevance
- Nursing roles include:
- Monitoring for GI bleeding signs (e.g., black stools, vomiting blood),
- Ensuring adherence to H. pylori eradication therapy,
- Educating on NSAID avoidance, smoking cessation, and stress management,
- Watching for surgical emergencies like perforation.
Pathological Changes in Gastritis – H. pylori Infection
Gastritis is the inflammation of the gastric mucosa, and when caused by Helicobacter pylori—a spiral-shaped, gram-negative bacillus—it leads to a chronic active form of gastritis that primarily affects the antrum of the stomach.
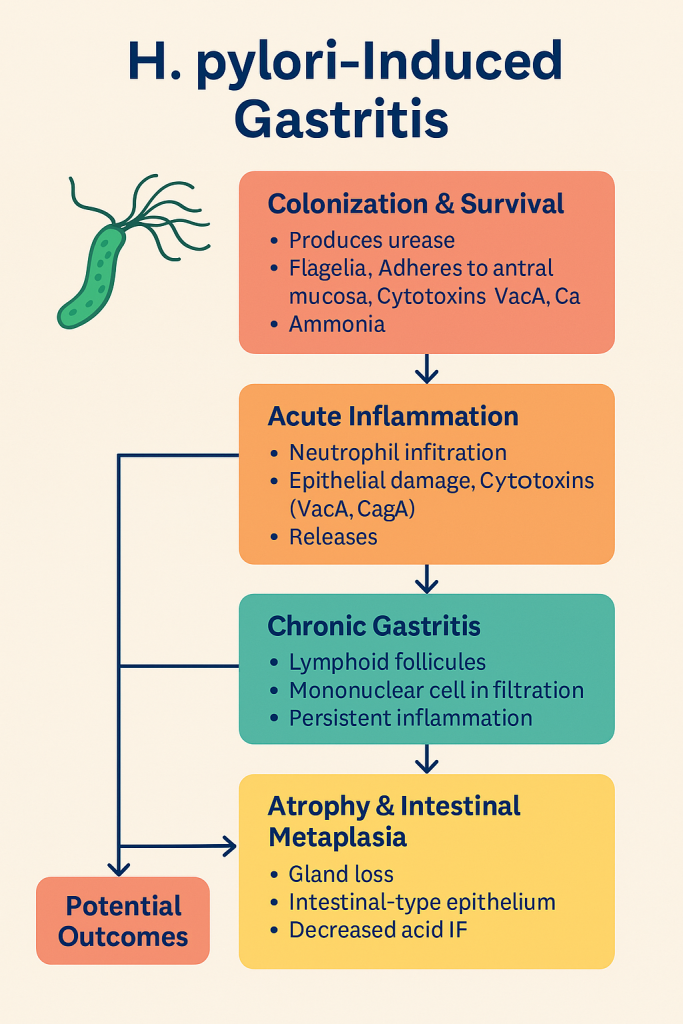
1. Colonization and Survival of H. pylori
- H. pylori survives in the harsh acidic environment of the stomach by:
- Producing urease, which breaks down urea into ammonia → neutralizes stomach acid.
- Using flagella to move beneath the mucus layer.
- Adhering to gastric epithelial cells, especially in the antrum.
- This creates a localized alkaline microenvironment, but simultaneously initiates mucosal damage.
2. Acute Inflammatory Reaction (Initial Mucosal Injury)
- H. pylori releases cytotoxins (e.g., CagA, VacA), proteases, and ammonia, causing:
- Disruption of tight junctions between epithelial cells.
- Destruction of surface mucus and epithelial cells.
- Direct epithelial injury.
- In response, the body recruits:
- Neutrophils, mononuclear cells, and lymphocytes into the lamina propria.
- Initiates acute-on-chronic inflammation.
3. Chronic Active Gastritis
- With persistent infection:
- Chronic lymphoplasmacytic infiltration develops in the lamina propria.
- Formation of lymphoid follicles (similar to MALT – mucosa-associated lymphoid tissue).
- Glandular atrophy and intestinal metaplasia may occur over time.
- This stage is typically asymptomatic or mildly symptomatic (dyspepsia).
4. Progression to Atrophic Gastritis and Intestinal Metaplasia
- Long-standing H. pylori infection leads to:
- Destruction of gastric glands (especially parietal and chief cells).
- Replacement by intestinal-type epithelium → intestinal metaplasia.
- Decreased acid and intrinsic factor secretion → risk of pernicious anemia.
- In some patients, this may progress to:
- Dysplasia → Gastric adenocarcinoma (intestinal type).
5. Potential Outcomes and Complications
- Peptic ulcer disease (most common complication, especially duodenal ulcers).
- Gastric MALT lymphoma (arising from lymphoid follicles).
- Chronic gastritis → Atrophy → Cancer sequence in susceptible individuals.
Summary in Chart Form
| Stage / Change | Key Pathological Features |
|---|---|
| Colonization & Survival | Urease production, flagella, adherence to epithelium |
| Acute Inflammation | Neutrophilic infiltration, epithelial damage, cytotoxin release |
| Chronic Gastritis | Lymphoid follicles, mononuclear infiltrate, persistent inflammation |
| Atrophy & Intestinal Metaplasia | Glandular loss, intestinal-type epithelium, reduced acid/IF secretion |
| Complications | Peptic ulcer, gastric cancer, MALT lymphoma |
Clinical and Nursing Relevance
- Symptoms: Epigastric pain, bloating, nausea, loss of appetite.
- Diagnosis:
- Urea breath test, stool antigen test, endoscopic biopsy, serology.
- Treatment:
- Triple therapy: PPI + Clarithromycin + Amoxicillin/Metronidazole.
- Eradication heals gastritis and prevents progression to ulcers or cancer.
- Nursing care includes:
- Ensuring compliance with full antibiotic course,
- Educating on avoiding NSAIDs, alcohol, and smoking,
- Monitoring for GI bleeding signs or anemia in chronic gastritis.
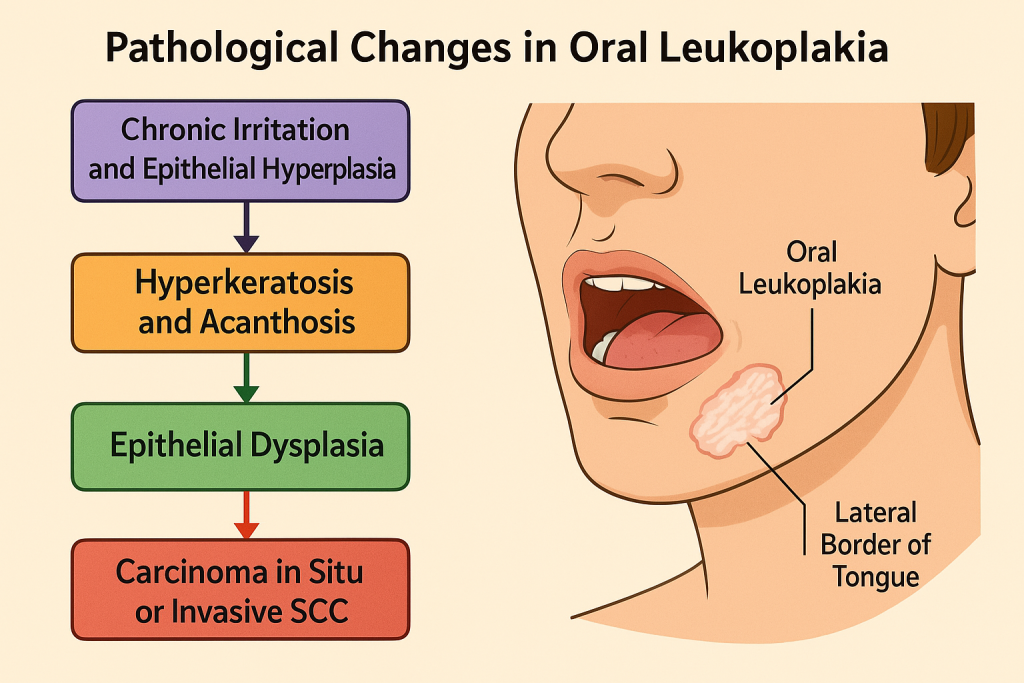
Pathological Changes in Oral Leukoplakia
Oral Leukoplakia is defined as a white patch or plaque in the oral cavity that cannot be scraped off and cannot be classified as any other diagnosable condition. It is considered a potentially malignant disorder of the oral mucosa and reflects a precancerous epithelial change.
Though not an infection in the classical sense, it is frequently associated with chronic irritants and microbial influences, including tobacco, alcohol, and HPV infection.
1. Initiation: Chronic Irritation and Epithelial Hyperplasia
- Primary risk factors:
- Tobacco use (smoked or chewed),
- Alcohol consumption (synergistic with tobacco),
- Mechanical irritation (sharp teeth, ill-fitting dentures),
- HPV (Human Papillomavirus), especially types 16 and 18,
- Candida albicans overgrowth (sometimes associated).
- Pathological Response:
- The squamous epithelium responds with hyperkeratosis (thickened keratin layer).
- Acanthosis: thickening of the spinous layer (stratum spinosum).
- Clinically appears as a non-removable white patch on the buccal mucosa, tongue, or floor of mouth.
2. Histopathological Spectrum: From Hyperplasia to Dysplasia
The severity of leukoplakia is assessed by the degree of epithelial dysplasia:
a) Benign Hyperkeratosis (No Dysplasia)
- Hyperorthokeratosis (increased orthokeratin without nuclei),
- Hyperparakeratosis (increased parakeratin with retained nuclei),
- No cellular atypia.
b) Epithelial Dysplasia (Precancerous)
- Loss of polarity of basal cells,
- Increased nuclear-to-cytoplasmic ratio,
- Pleomorphism, hyperchromatism,
- Abnormal mitotic figures,
- Dysplastic changes may extend from mild → moderate → severe.
🔄 3. Advanced Stage: Carcinoma in Situ or Invasive SCC
- In high-risk lesions:
- The full thickness of the epithelium shows dysplastic changes → Carcinoma in situ.
- Breach of the basement membrane leads to invasive squamous cell carcinoma (SCC).
- Most common sites for malignant transformation:
- Lateral border of tongue, floor of mouth, soft palate.
🧬 4. Immunological and Molecular Pathogenesis
- Chronic irritation and HPV-related oncogenesis lead to:
- Mutation in tumor suppressor genes (e.g., p53),
- Activation of oncogenes,
- Inhibition of apoptosis,
- Promotion of uncontrolled epithelial proliferation.
🧾 Summary in Chart Form
| Stage / Change | Key Pathological Features |
|---|---|
| Chronic Irritation | Tobacco, alcohol, trauma, HPV; leads to epithelial hyperplasia |
| Hyperkeratosis & Acanthosis | Thickened keratin and epithelial layers, white patch |
| Epithelial Dysplasia | Nuclear atypia, pleomorphism, abnormal mitosis |
| Carcinoma in Situ / Invasion | Full-thickness atypia or basement membrane invasion |
| Molecular Changes | p53 mutation, oncogene activation, loss of cell cycle control |
✨ Clinical and Nursing Relevance
- Symptoms: White patch that persists >2 weeks, usually painless, but may feel rough.
- Diagnosis: Clinical inspection + biopsy for histopathology.
- Management:
- Stop tobacco/alcohol use.
- Surgical excision for dysplastic lesions.
- Regular surveillance for malignant transformation.
- Nursing focus:
- Oral health education, tobacco cessation support,
- Monitor for suspicious mucosal changes,
- Encourage biopsy and follow-up care.
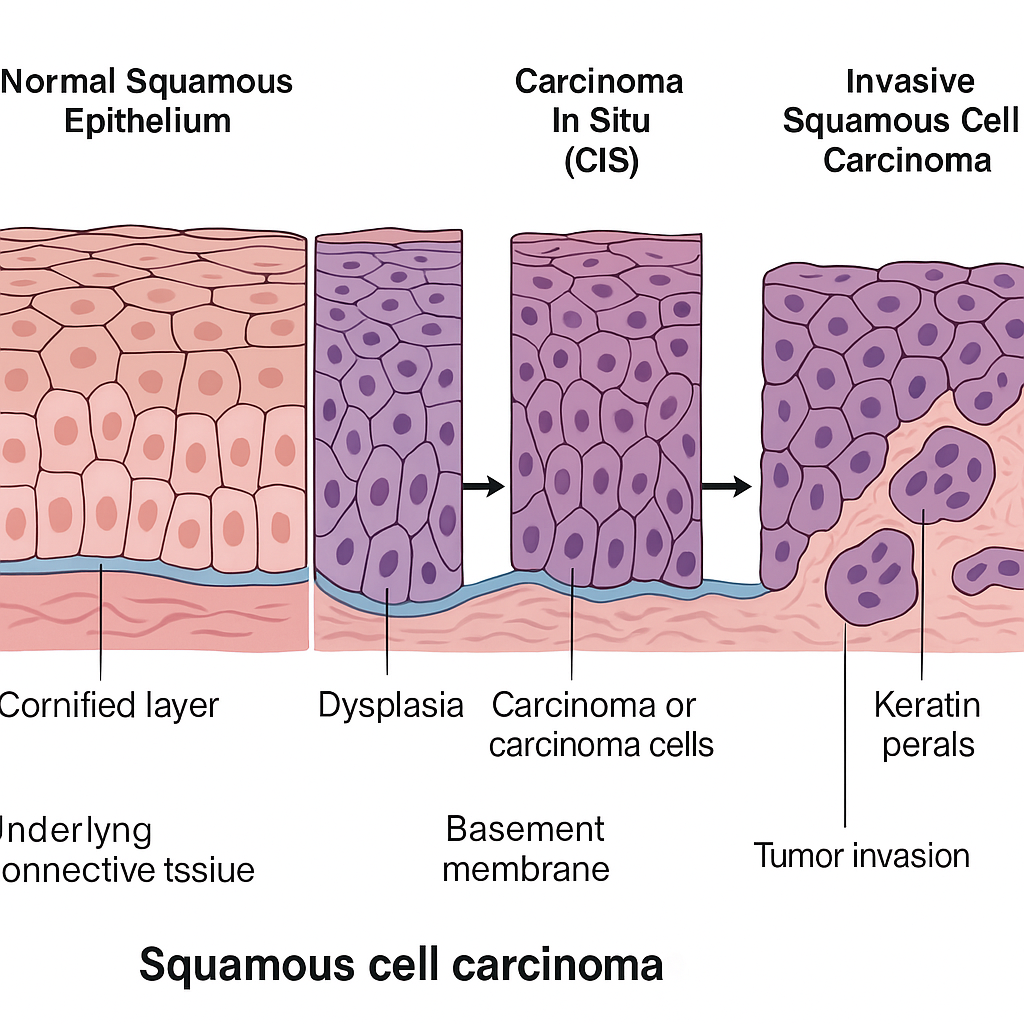
Squamous Cell Carcinoma – Pathological Changes
🧠✨Let’s explore the microscopic-to-macroscopic transformation of squamous cell carcinoma (SCC), beginning from the normal squamous epithelium to full-blown invasive cancer.
🔬 1. Introduction to Squamous Cell Carcinoma (SCC):
Squamous cell carcinoma is a malignant neoplasm arising from squamous epithelial cells, which are flat, scale-like cells typically found lining the skin, respiratory tract, oral cavity, esophagus, cervix, and other areas.
It is the second most common form of skin cancer, but can also arise in internal organs lined by squamous epithelium (e.g., lung, esophagus, cervix).
🔄 2. Sequence of Pathological Changes (Histogenesis):
🧩 A. Normal Squamous Epithelium:
- Well-organized layers:
- Basal layer (mitotically active)
- Spinous layer
- Granular layer
- Cornified (keratin) layer
- Cells show polarity, maturation, and adhesion.
🔁 B. Squamous Metaplasia:
- Reversible change where non-squamous epithelium transforms into squamous epithelium due to chronic irritation or inflammation (e.g., smoking, HPV infection).
⚠️ C. Dysplasia (Premalignant Change):
- Disordered growth and maturation of squamous cells.
- Loss of polarity, nuclear atypia, increased mitotic figures, and basement membrane still intact.
- Graded as mild, moderate, or severe dysplasia.
🚨 D. Carcinoma In Situ (CIS):
- Severe dysplasia involving the full thickness of the epithelium.
- No invasion beyond the basement membrane yet.
- High risk of progression to invasive carcinoma.
🧨 E. Invasive Squamous Cell Carcinoma:
- Malignant cells breach the basement membrane and invade underlying tissues.
- Irregular nests, cords, and sheets of squamous cells with:
- Keratin pearls
- Intercellular bridges
- Hyperchromatic nuclei
- Prominent nucleoli
- Evidence of vascular and lymphatic invasion may be seen.
📊 3. Morphological Features (Macroscopic and Microscopic):
🔍 Gross (Macroscopic) Appearance:
- May appear as:
- Ulcerated lesion with raised everted edges.
- Exophytic mass (cauliflower-like growth).
- Infiltrative indurated plaque.
- Commonly affects sun-exposed skin, oral mucosa, cervix, and lungs.
🧫 Microscopic (Histological) Features:
- Polygonal squamous cells with eosinophilic cytoplasm.
- Keratin pearl formation (concentric layers of keratin).
- Intercellular bridges (desmosomes between cells).
- Varying degrees of differentiation:
- Well-differentiated: More keratin, visible pearls.
- Poorly differentiated: Less keratin, more pleomorphism.
🧠 4. Molecular Pathogenesis Highlights:
- UV radiation (in cutaneous SCC) → DNA damage, p53 mutation.
- HPV infection (especially types 16, 18 in cervical SCC).
- Smoking and alcohol (oral and esophageal SCC).
- Chronic inflammation and irritation (e.g., lichen sclerosis, actinic keratosis).
🔄 Summary Chart of Changes (Quick Reference):
| Stage | Changes |
|---|---|
| Normal | Orderly squamous layers, no atypia |
| Metaplasia | Non-squamous epithelium becomes squamous due to irritation |
| Dysplasia | Cellular atypia, disordered layers, intact basement membrane |
| Carcinoma In Situ | Full-thickness atypia, basement membrane intact |
| Invasive SCC | Malignant cells invade below basement membrane, keratin pearls, atypia |
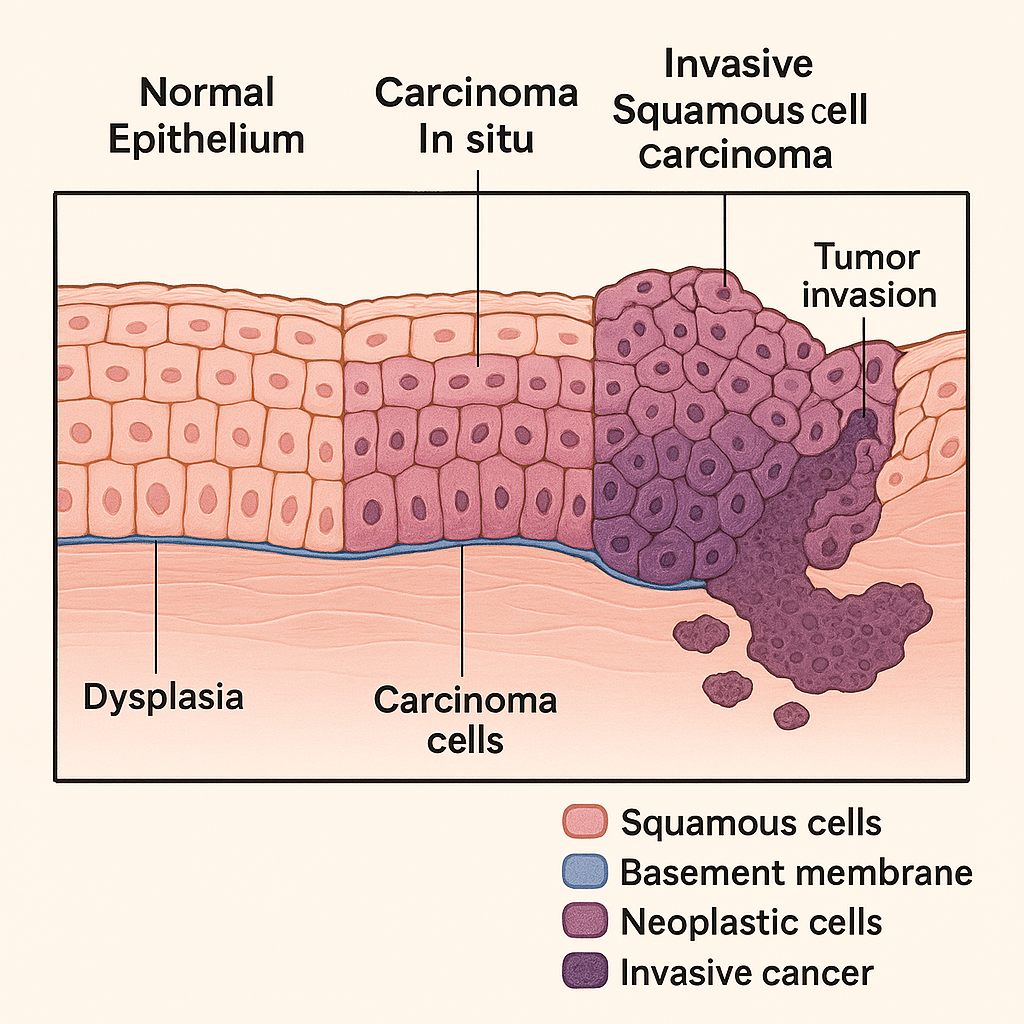
Pathological Changes in Esophageal Cancer
Esophageal cancer is a malignant neoplasm of the esophagus characterized by progressive dysplastic changes of the epithelial lining, culminating in invasive carcinoma. Two major histological types exist:
- Squamous Cell Carcinoma (SCC) – arises from the squamous epithelium of the upper/middle esophagus.
- Adenocarcinoma – arises from glandular metaplasia (Barrett’s esophagus) in the lower esophagus.
🔬 1. Histogenesis and Pathological Progression
The pathological evolution of esophageal cancer typically follows a multistep process that begins with chronic mucosal injury and ends in invasive malignancy:
🔹 A. Chronic Irritation or Mucosal Injury
- Causative Factors:
- Smoking, alcohol (for SCC)
- Chronic gastroesophageal reflux disease (GERD) and Barrett’s esophagus (for adenocarcinoma)
- Hot beverages, achalasia, caustic strictures
- Leads to persistent inflammation, oxidative damage, and mucosal cell turnover
🔹 B. Metaplasia
- In adenocarcinoma: Chronic GERD → intestinal metaplasia of distal esophagus (Barrett’s Esophagus)
- Normal squamous epithelium is replaced by columnar epithelium with goblet cells
- In SCC: Chronic irritation causes atypical squamous hyperplasia
🔹 C. Dysplasia (Low → High Grade)
- Disordered epithelial cell maturation
- Loss of polarity, nuclear enlargement, increased mitotic figures
- Low-grade dysplasia progresses to high-grade dysplasia:
- Intact basement membrane but severe cytologic atypia
- Marker of precancerous transformation
🔹 D. Carcinoma In Situ
- Entire thickness of epithelium is involved with dysplastic changes
- Still no invasion through the basement membrane
- High risk of progression to invasive carcinoma
🔹 E. Invasive Esophageal Carcinoma
- Malignant cells invade past the basement membrane into submucosa and muscularis
- Irregular nests or glands (depending on type) infiltrate deep layers
- May involve regional lymphatics, blood vessels, and metastasize to distant organs like liver, lungs, or bones
🧫 2. Histopathological Features by Type
📘 A. Squamous Cell Carcinoma (SCC)
- Arises in middle third of esophagus
- Microscopy:
- Malignant squamous cells with intercellular bridges
- Keratin pearl formation
- Dense, eosinophilic cytoplasm
- Often associated with inflammation, necrosis, and fibrosis
📘 B. Adenocarcinoma
- Typically arises in lower third (Barrett’s esophagus background)
- Microscopy:
- Gland-forming tumor cells
- Infiltrative, mucin-producing malignant epithelium
- Frequently shows intestinal-type differentiation
👁️ 3. Gross (Macroscopic) Appearance
- Polypoid/exophytic mass: protrudes into lumen
- Ulcerative lesion: central necrosis with rolled edges
- Infiltrative type: wall thickening, stricture formation
- Advanced tumors: may encircle esophagus → dysphagia (progressive, starting with solids)
🔬 4. Molecular & Genetic Changes
- p53 mutation (common in both SCC and adenocarcinoma)
- Cyclin D1, EGFR, HER2 amplification (more in adenocarcinoma)
- CDKN2A loss, NOTCH1, and SOX2 mutations in SCC
📊 Summary Table – Pathological Evolution
| Stage | Description |
|---|---|
| Normal Epithelium | Stratified squamous epithelium in upper/mid esophagus; columnar metaplasia in lower esophagus (Barrett’s) |
| Metaplasia | Squamous → columnar (adenocarcinoma), atypical hyperplasia (SCC) |
| Dysplasia | Disordered growth, nuclear atypia, increased mitosis |
| Carcinoma in Situ | Full-thickness epithelial dysplasia, no invasion |
| Invasive Cancer | Malignant cells breach basement membrane, invade deeper layers |
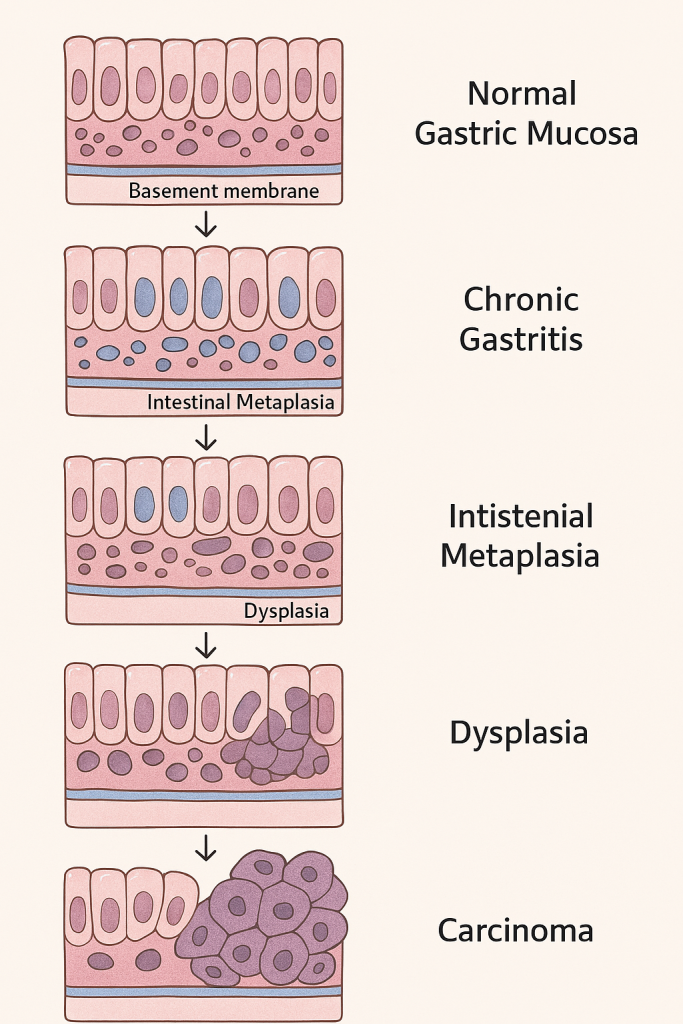
🧬 Pathological Changes in Gastric Cancer
Gastric cancer, also known as stomach cancer, is a malignant neoplasm originating primarily from the gastric mucosa. It is the fifth most common cancer worldwide and the third leading cause of cancer-related deaths. The most frequent type is adenocarcinoma, but others include lymphomas, GISTs (gastrointestinal stromal tumors), and neuroendocrine tumors.
Let’s explore the sequential pathological changes, from normal gastric mucosa to invasive cancer, with a focus on adenocarcinoma.
🔄 1. Histogenesis of Gastric Adenocarcinoma (Correa’s Cascade)
The development of gastric adenocarcinoma typically follows a multistep process, especially in intestinal-type carcinoma, often described by the Correa pathway:
🔹 A. Normal Gastric Mucosa
- Comprised of columnar epithelial cells with specialized gastric glands (chief, parietal, mucous, G cells).
- Maintains tight cell junctions, polarity, and low mitotic activity.
🔹 B. Chronic Gastritis
- Persistent H. pylori infection, autoimmunity, or dietary carcinogens (e.g., nitrosamines) cause:
- Mucosal inflammation
- Neutrophil infiltration
- Lymphoid follicles
- Glandular atrophy may develop over time.
🔹 C. Intestinal Metaplasia
- Native gastric epithelium transforms into intestinal-type epithelium (goblet cells, absorptive cells, Paneth cells).
- Seen as a defensive adaptation but is premalignant.
🔹 D. Dysplasia (Intraepithelial Neoplasia)
- Low-grade dysplasia: Architectural distortion, mild nuclear atypia.
- High-grade dysplasia: Loss of polarity, marked pleomorphism, hyperchromasia, frequent mitoses.
- The basement membrane remains intact.
🔹 E. Invasive Gastric Adenocarcinoma
- Malignant epithelial cells invade beyond the basement membrane into:
- Lamina propria
- Muscularis mucosae
- Submucosa
- Muscularis propria
- Histological types:
- Intestinal type: Gland-forming, better differentiated, solid mass.
- Diffuse type: Poorly differentiated, signet ring cells, infiltrative, rigid stomach wall (linitis plastica).
🧫 2. Microscopic Features by Histological Type
📘 Intestinal Type Adenocarcinoma
- Forms gland-like structures
- Associated with H. pylori, chronic gastritis, and atrophy
- Nuclear atypia, hyperchromasia, glandular crowding
📘 Diffuse Type Adenocarcinoma
- No gland formation
- Signet ring cells with mucin pushing nuclei to periphery
- Loss of E-cadherin expression
- Thickened gastric wall, “leather bottle” appearance
👁️ 3. Macroscopic (Gross) Types of Gastric Cancer
(Borrmann Classification for Advanced Gastric Cancer)
| Type | Appearance |
|---|---|
| Type I | Polypoid/fungating mass |
| Type II | Ulcerated lesion with raised edges |
| Type III | Ulcerated and infiltrative |
| Type IV | Diffusely infiltrative (linitis plastica) |
🧬 4. Molecular and Genetic Alterations
- H. pylori → inflammation → DNA damage
- p53 mutations, APC, KRAS, E-cadherin (CDH1) loss in diffuse type
- Microsatellite instability (MSI) and Epstein-Barr virus (EBV) linked to specific subtypes
📊 Summary of Pathological Changes in Gastric Cancer
| Stage | Changes |
|---|---|
| Normal Gastric Epithelium | Intact glandular epithelium |
| Chronic Gastritis | Inflammatory infiltrate, gland loss |
| Intestinal Metaplasia | Goblet cells replace gastric epithelium |
| Dysplasia | Nuclear atypia, architectural distortion |
| Carcinoma In Situ | Severe dysplasia with intact basement membrane |
| Invasive Carcinoma | Malignant cells invade mucosa and deeper layers |
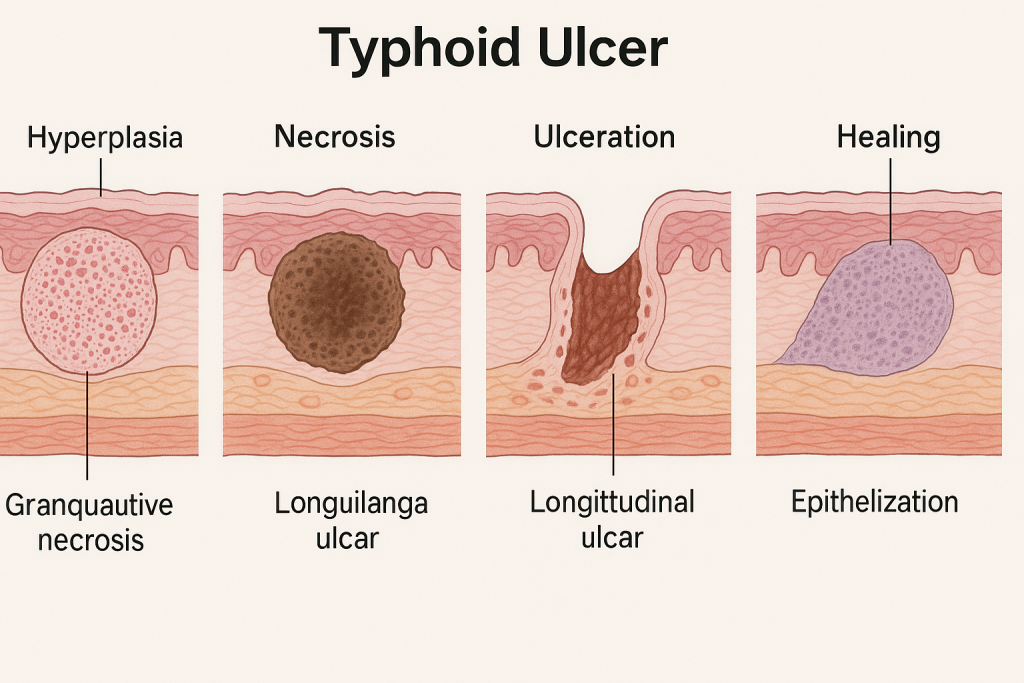
🧬 Pathological Changes in Typhoid Ulcer (Enteric Fever Ulceration)
Typhoid fever, caused by Salmonella typhi (a gram-negative bacillus), leads to a systemic infection affecting various organs, especially the intestinal lymphoid tissues, particularly in the ileum. One of the hallmark lesions of this disease is the “typhoid ulcer”, which is a result of necrosis of lymphoid tissue followed by ulceration of the overlying mucosa.
Let’s explore the pathological progression of these ulcers:
🔬 1. Initial Site of Pathology: Peyer’s Patches
- Peyer’s patches are aggregations of lymphoid tissue in the terminal ileum.
- These are the primary sites where S. typhi invades and multiplies.
- Bacteria penetrate M cells in the intestinal epithelium → engulfed by macrophages → spread through lymphatics and bloodstream (bacteremia).
🔄 2. Stages of Pathological Changes in Typhoid Ulcers
🔹 Stage 1: Hyperplasia of Peyer’s Patches
- Peyer’s patches enlarge due to lymphoid hyperplasia and intense mononuclear cell infiltration.
- Seen around the first week of illness.
- Grossly: plaques become elevated and swollen, with dusky appearance.
🔹 Stage 2: Necrosis
- Central portion of the hyperplastic patch undergoes coagulative necrosis.
- Due to endotoxins and ischemia caused by local thrombosis.
- Third week: necrosis reaches overlying mucosa.
🔹 Stage 3: Ulceration
- Necrotic tissue sloughs off → forms longitudinal, oval ulcers.
- Ulcers are:
- Oriented along the long axis of the bowel (diagnostic feature)
- Margins are clean and not undermined
- Base contains granulation tissue, necrotic debris, and inflammatory cells
🔹 Stage 4: Healing
- Begins by the fourth week.
- Granulation tissue forms, followed by fibrosis and epithelial regeneration.
- In uncomplicated cases, ulcers heal without scarring.
🧫 Microscopic Features
- Necrosis of mucosa, submucosa, and lymphoid tissue
- Inflammatory infiltrate: mononuclear cells (macrophages, plasma cells, lymphocytes)
- Thrombosis of small blood vessels
- Granulomatous inflammation with prominent macrophages (typhoid cells)
⚠️ Complications of Typhoid Ulcer
- Intestinal perforation (most severe, life-threatening)
- Massive intestinal hemorrhage
- Strictures and adhesions (rare)
- Peritonitis if contents leak from perforated ulcer
📊 Summary of Pathological Changes
| Stage | Description |
|---|---|
| Hyperplasia | Swelling of Peyer’s patches, mononuclear infiltration |
| Necrosis | Central coagulative necrosis due to bacterial toxins and thrombosis |
| Ulceration | Sloughing off necrotic tissue; longitudinal ulcers with clean, regular margins |
| Healing | Regeneration of mucosa with granulation tissue and fibrosis |
🧠 Clinical Correlation
- Abdominal pain, diarrhea, and GI bleeding are signs of ulceration.
- Sudden abdominal rigidity may indicate perforation – a surgical emergency.
- Diagnosis is supported by Widal test, blood cultures, and bone marrow cultures.
🧬 Pathological Changes in Crohn’s Disease
Crohn’s disease is a chronic granulomatous inflammatory bowel disease (IBD) that can affect any part of the gastrointestinal tract, from the mouth to the anus, but most commonly involves the terminal ileum and colon. It is characterized by transmural inflammation, skip lesions, and non-caseating granulomas, setting it apart from ulcerative colitis.
Let’s explore the pathological evolution and tissue-level changes in Crohn’s disease:
🔍 1. Gross (Macroscopic) Pathological Features
🔹 A. Segmental Distribution (Skip Lesions)
- The disease affects patchy areas of the bowel, leaving normal segments in between.
- Most commonly involved: terminal ileum, ileocecal region, colon
🔹 B. Thickened Bowel Wall
- Due to chronic inflammation, fibrosis, and hypertrophy of muscularis propria
- Leads to narrowed lumen and stricture formation
🔹 C. Cobblestone Appearance
- Longitudinal ulcers intersected by transverse edematous mucosal ridges
- Gives a cobblestone-like pattern on gross inspection
🔹 D. Fissures and Fistulas
- Deep ulcers may extend transmurally to form:
- Sinus tracts
- Fistulas (e.g., entero-enteric, rectovaginal, perianal)
- Abscesses
🔹 E. Creeping Fat
- Mesenteric fat wraps around the serosa of the bowel (mesenteric border) due to chronic inflammation
🔬 2. Microscopic (Histopathological) Features
🧫 A. Transmural Inflammation
- Involves mucosa, submucosa, muscularis propria, and serosa
- Lymphoid aggregates, crypt abscesses, and fibrosis may be seen in all layers
🧫 B. Non-Caseating Granulomas
- Hallmark of Crohn’s disease (in ~35–50% cases)
- Composed of epithelioid histiocytes, multinucleated giant cells, surrounded by lymphocytes
- Not associated with necrosis (non-caseating)
🧫 C. Mucosal Changes
- Focal crypt distortion, crypt abscesses, and atrophy
- Ulcers: longitudinal, serpiginous
- Goblet cell depletion may be mild or absent
🔁 3. Evolution of Crohn’s Lesion
- Early stage: Aphthous ulcers in mucosa
- Progression: Ulcers deepen, become linear and transmural
- Fibrosis: Healing leads to fibrostenotic areas
- Chronic stage: Fistula formation, strictures, bowel obstruction
⚠️ 4. Complications of Crohn’s Disease
| Complication | Explanation |
|---|---|
| Fistula formation | Due to transmural ulceration |
| Intestinal obstruction | Fibrosis and stricture narrowing lumen |
| Perforation | Rare, but possible with deep ulceration |
| Malabsorption | Especially in ileal involvement (vitamin B12, bile salts) |
| Perianal disease | Abscesses, fistulas, ulcers |
| Colon cancer | Increased risk after long-standing disease |
📊 Summary Table of Pathological Features
| Feature | Crohn’s Disease |
|---|---|
| Site | Any GI tract (commonly terminal ileum) |
| Distribution | Patchy (skip lesions) |
| Depth of inflammation | Transmural |
| Granulomas | Non-caseating granulomas |
| Ulcers | Longitudinal, serpiginous |
| Cobblestone appearance | Yes |
| Fistula and stricture | Common |
| Mesenteric fat involvement | Creeping fat |
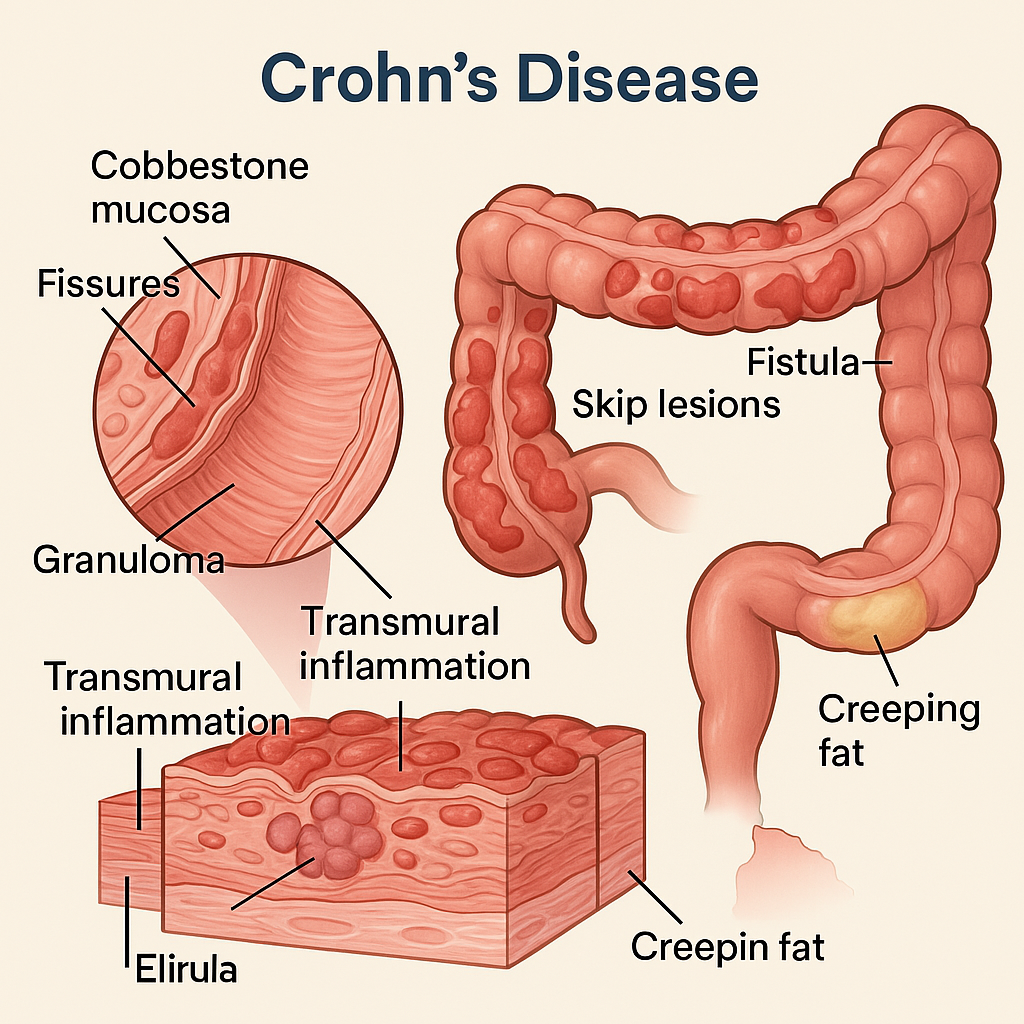
🧬 Pathological Changes in Ulcerative Colitis (UC)
Ulcerative colitis (UC) is a chronic inflammatory bowel disease (IBD) characterized by superficial, continuous inflammation of the colonic mucosa, starting from the rectum and extending proximally in a continuous manner through the colon. Unlike Crohn’s disease, UC is limited to the colon and rectum and involves only the mucosa and submucosa.
Let’s explore the pathological progression of UC and its defining features:
🔍 1. Gross (Macroscopic) Pathological Features
🔹 A. Continuous Involvement
- UC always starts at the rectum (proctitis) and may extend proximally to involve:
- Sigmoid colon → descending colon → transverse colon → up to the cecum
- No skip lesions (in contrast to Crohn’s)
🔹 B. Red, Granular, Friable Mucosa
- Inflamed mucosa becomes edematous, hemorrhagic, and friable (bleeds easily)
- Loss of normal mucosal folds due to inflammation
🔹 C. Pseudopolyps
- Islands of regenerating mucosa between ulcerated areas
- Appear as projecting polyp-like structures, but are non-neoplastic
🔹 D. Ulcers
- Broad-based, superficial ulcers confined to the mucosa
- May coalesce, leading to large denuded areas
🔬 2. Microscopic (Histopathological) Features
🧫 A. Inflammation Confined to Mucosa and Submucosa
- Unlike Crohn’s, transmural involvement is absent
- Diffuse infiltration of inflammatory cells (neutrophils, lymphocytes, plasma cells) in lamina propria
🧫 B. Crypt Architectural Distortion
- Crypts become branched, shortened, and irregular
- Goblet cell depletion due to chronic damage
🧫 C. Crypt Abscesses
- Neutrophils accumulate within the crypt lumina, forming crypt abscesses
- One of the histological hallmarks of active UC
🧫 D. Ulceration
- Limited to mucosa
- Overlying mucosal layer is denuded, with granulation tissue in the base
🔁 3. Stages of Pathological Evolution
| Stage | Description |
|---|---|
| Early | Rectal inflammation with mild mucosal edema and hyperemia |
| Progression | Extension proximally, crypt distortion, goblet cell loss |
| Active phase | Ulcers, crypt abscesses, heavy inflammatory infiltrate |
| Chronic phase | Mucosal atrophy, pseudopolyps, architectural changes |
⚠️ 4. Complications of Ulcerative Colitis
| Complication | Description |
|---|---|
| Toxic megacolon | Acute dilation with risk of perforation |
| Perforation | Rare but severe |
| Massive hemorrhage | From ulcerated mucosa |
| Colorectal carcinoma | Increased risk with chronic UC (>10 years) |
| Strictures (rare) | Usually due to healing and fibrosis |
| Primary sclerosing cholangitis | Extraintestinal manifestation involving bile ducts |
📊 Summary of Key Pathological Differences (vs. Crohn’s)
| Feature | Ulcerative Colitis | Crohn’s Disease |
|---|---|---|
| Area involved | Colon & rectum only | Mouth to anus (especially ileum) |
| Distribution | Continuous | Skip lesions |
| Depth of involvement | Mucosa and submucosa | Transmural |
| Ulcers | Broad-based, superficial | Linear, deep |
| Pseudopolyps | Common | Less common |
| Granulomas | Absent | Present (non-caseating) |
| Cobblestone mucosa | No | Yes |
| Fistula/stricture formation | Rare | Common |
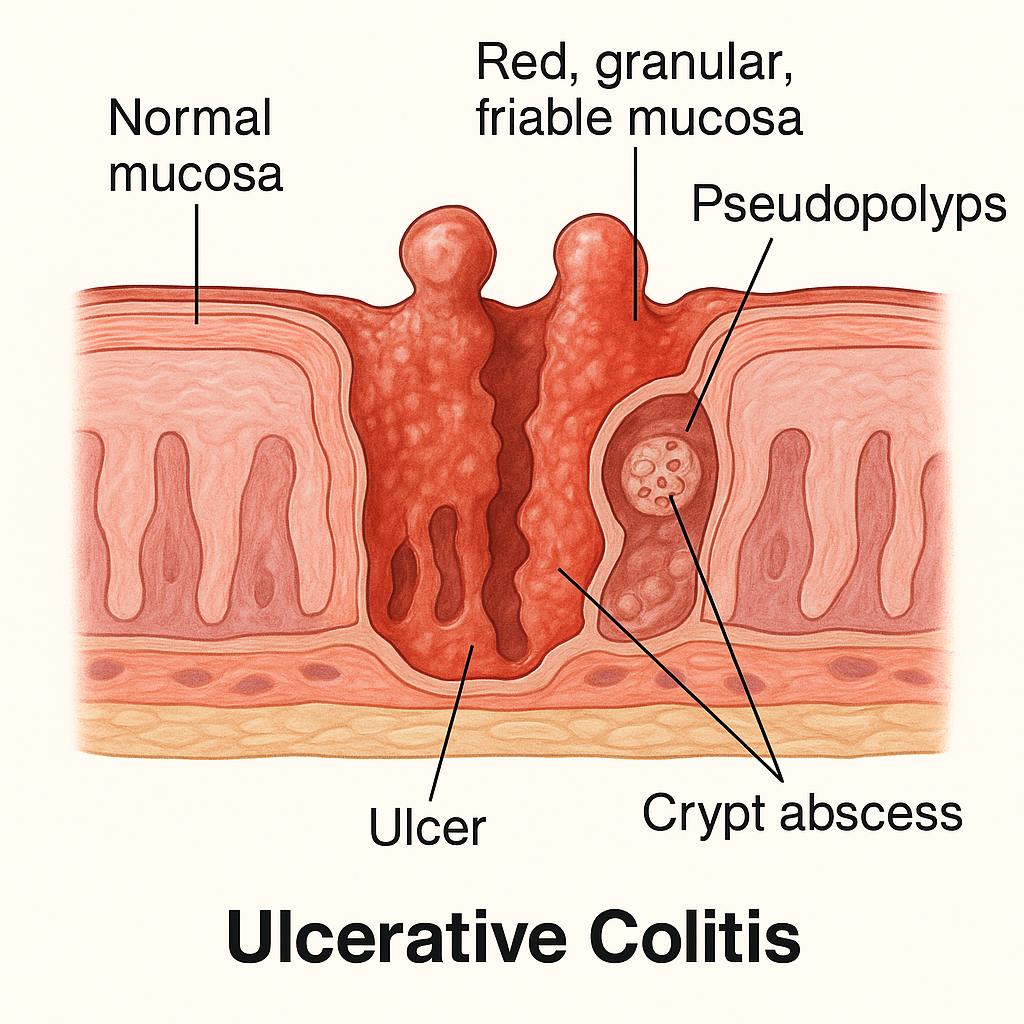
🧬 Pathological Changes in Colorectal Cancer (CRC)
Colorectal cancer (CRC) is a malignant neoplasm arising from the epithelial lining of the colon or rectum. Most CRCs are adenocarcinomas, developing from pre-existing adenomatous polyps through a multistep sequence of genetic and morphological changes. This is classically known as the adenoma-carcinoma sequence (Vogelstein model).
Let’s explore the pathological progression, histological features, and molecular changes in colorectal cancer.
🔬 1. Histogenesis – Adenoma-Carcinoma Sequence
🔹 A. Normal Epithelium
- Lined by simple columnar epithelium with goblet cells
- Regulated cell proliferation and apoptosis
- Normal architecture of crypts of Lieberkühn
🔹 B. Early Adenoma (Polyp Formation)
- Begins with a mutation in the APC gene (tumor suppressor)
- Leads to excessive cell proliferation in the crypt base
- Forms a tubular or villous adenoma
- Tubular adenoma: rounded, gland-like structures
- Villous adenoma: finger-like projections, more malignant potential
🔹 C. Advanced Adenoma
- Accumulation of additional mutations (e.g., KRAS oncogene)
- Displays increased dysplasia:
- Loss of cellular polarity
- Nuclear hyperchromasia
- Increased mitotic activity
- High-grade dysplasia is a direct precursor to carcinoma
🔹 D. Invasive Adenocarcinoma
- Final hit: mutation or loss of p53 tumor suppressor gene
- Tumor invades through the basement membrane into:
- Submucosa
- Muscularis propria
- Serosa and beyond
- May metastasize to regional lymph nodes, liver, lungs, and bones
🧫 2. Histological Features of Colorectal Adenocarcinoma
📘 Microscopic Appearance:
- Infiltrative, irregular glandular structures
- Malignant epithelial cells with:
- Large, hyperchromatic nuclei
- Prominent nucleoli
- Loss of mucin in some poorly differentiated forms
- Desmoplastic stromal reaction (fibrous tissue surrounding cancer)
- Dirty necrosis within gland lumens (seen especially in colorectal origin)
🔍 3. Gross (Macroscopic) Appearance
🔹 Left-Sided Lesions (Descending colon, sigmoid, rectum):
- Annular (napkin-ring) constricting growth
- Leads to intestinal obstruction
- Often associated with change in bowel habits and hematochezia (fresh blood)
🔹 Right-Sided Lesions (Cecum and ascending colon):
- Exophytic, polypoid masses
- Less likely to cause obstruction
- Symptoms: iron-deficiency anemia, fatigue, occult bleeding
🧬 4. Molecular Pathways of CRC Development
| Pathway | Key Genes Involved | Notes |
|---|---|---|
| Chromosomal instability | APC, KRAS, p53 | Classic adenoma-carcinoma sequence |
| Microsatellite instability | MLH1, MSH2, MSH6 (MMR genes) | Lynch syndrome (HNPCC) |
| CpG island methylator phenotype (CIMP) | Hypermethylation of promoter regions | Often overlaps with MSI pathway |
⚠️ 5. Complications of Colorectal Cancer
- Bowel obstruction (especially left-sided)
- Perforation and peritonitis
- Liver metastasis (via portal vein)
- Anemia (especially in right-sided cancers)
- Weight loss, cachexia
- Paraneoplastic syndromes
📊 Summary Table of Pathological Changes
| Stage | Changes |
|---|---|
| Normal Epithelium | Healthy columnar cells, intact architecture |
| Adenomatous Polyp | Glandular hyperplasia, nuclear atypia, architectural changes |
| High-Grade Dysplasia | Loss of polarity, mitotic activity, prominent nuclei |
| Invasive Adenocarcinoma | Penetration of muscularis, stromal invasion, gland formation disrupted |
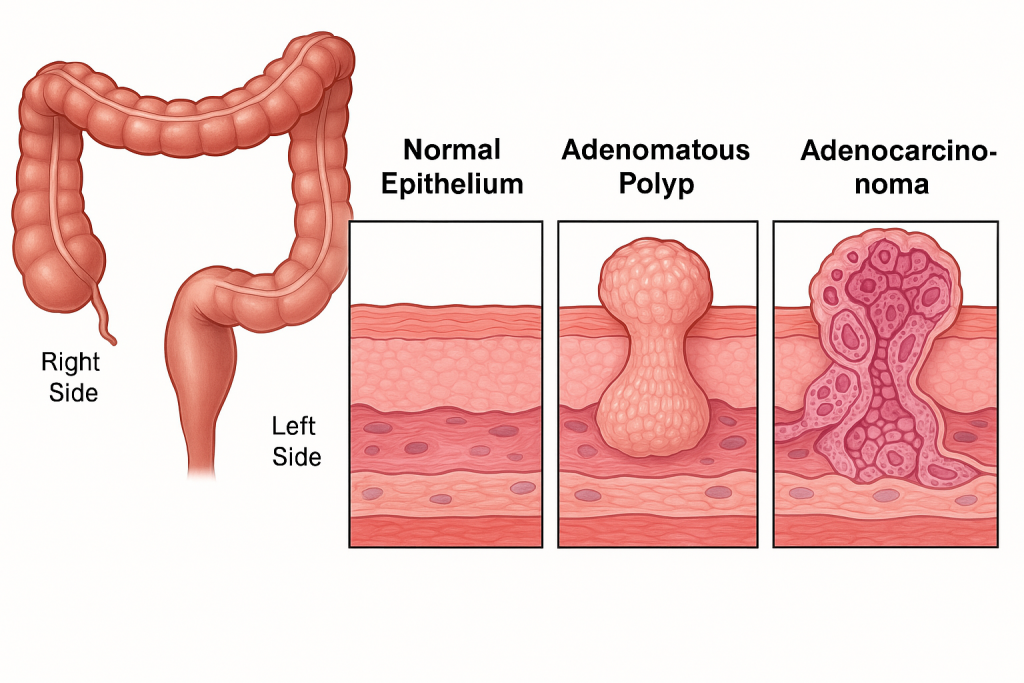
Liver, Gall Bladder and Pancreas
🧬 Pathological Changes in Hepatitis
(A detailed and academic explanation with structured progression and clinical insight)
Hepatitis refers to inflammation of the liver, which can result from viral infections, autoimmune disorders, toxins (e.g., alcohol, drugs), or metabolic diseases. The most common and studied forms are viral hepatitis, caused by hepatitis viruses A, B, C, D, and E.
Regardless of etiology, the pathological changes in hepatitis share common stages involving hepatocyte injury, inflammation, necrosis, and possibly fibrosis or cirrhosis if chronic.
🔬 1. Morphological Classification Based on Duration
🔹 Acute Hepatitis
- Short-term inflammation (usually <6 months)
- Caused by: HAV, HEV, HBV (sometimes), drugs, toxins
🔹 Chronic Hepatitis
- Persistent inflammation for >6 months
- Caused mainly by: HBV, HCV, HDV, autoimmune hepatitis
🔄 2. Pathological Stages of Hepatitis (Histological Progression)
🔹 A. Hepatocellular Injury
- Ballooning degeneration: swollen hepatocytes with pale cytoplasm due to water accumulation
- Apoptosis of hepatocytes: shows as acidophilic Councilman bodies
- Necrosis: focal or confluent hepatocellular necrosis (more severe in toxic hepatitis)
🔹 B. Inflammatory Infiltration
- Involves portal tracts and lobules
- Mostly lymphocytes (especially in viral hepatitis)
- In autoimmune hepatitis: plasma cells predominant
- In fulminant cases: neutrophils and extensive inflammation
🔹 C. Interface Hepatitis (Piecemeal Necrosis)
- In chronic hepatitis, inflammatory cells spill over from the portal tracts into adjacent parenchyma
- Associated with fibrosis progression
- A key diagnostic feature for chronic viral or autoimmune hepatitis
🔹 D. Fibrosis and Cirrhosis (Chronic Stage)
- Persistent inflammation stimulates stellate cells → deposition of collagen
- Leads to bridging fibrosis connecting portal tracts and central veins
- Eventually forms regenerative nodules → cirrhosis
🧫 3. Specific Histopathological Features by Etiology
| Type | Key Histological Features |
|---|---|
| Acute Viral Hepatitis | Ballooning degeneration, spotty necrosis, Councilman bodies, mononuclear infiltration |
| Chronic Hepatitis B | Ground-glass hepatocytes (due to HBsAg), interface hepatitis, fibrosis |
| Chronic Hepatitis C | Lymphoid aggregates in portal areas, bile duct injury, steatosis |
| Autoimmune Hepatitis | Dense portal inflammation with plasma cells, interface hepatitis |
| Alcoholic Hepatitis | Mallory bodies, neutrophils, ballooning, perivenular fibrosis |
⚠️ 4. Complications of Chronic Hepatitis
| Complication | Explanation |
|---|---|
| Cirrhosis | End-stage liver fibrosis with regenerative nodules |
| Portal hypertension | Due to architectural distortion |
| Liver failure | Decreased synthetic/metabolic capacity |
| Hepatocellular carcinoma (HCC) | Especially in HBV/HCV-related cirrhosis |
📊 Summary of Pathological Evolution in Hepatitis
| Stage | Microscopic Features |
|---|---|
| Hepatocyte injury | Ballooning, apoptosis, necrosis |
| Inflammatory phase | Mononuclear infiltration (portal/lobular) |
| Interface hepatitis | Inflammation at limiting plate |
| Fibrosis | Collagen deposition, starts periportal |
| Cirrhosis | Bridging fibrosis, nodular regeneration |
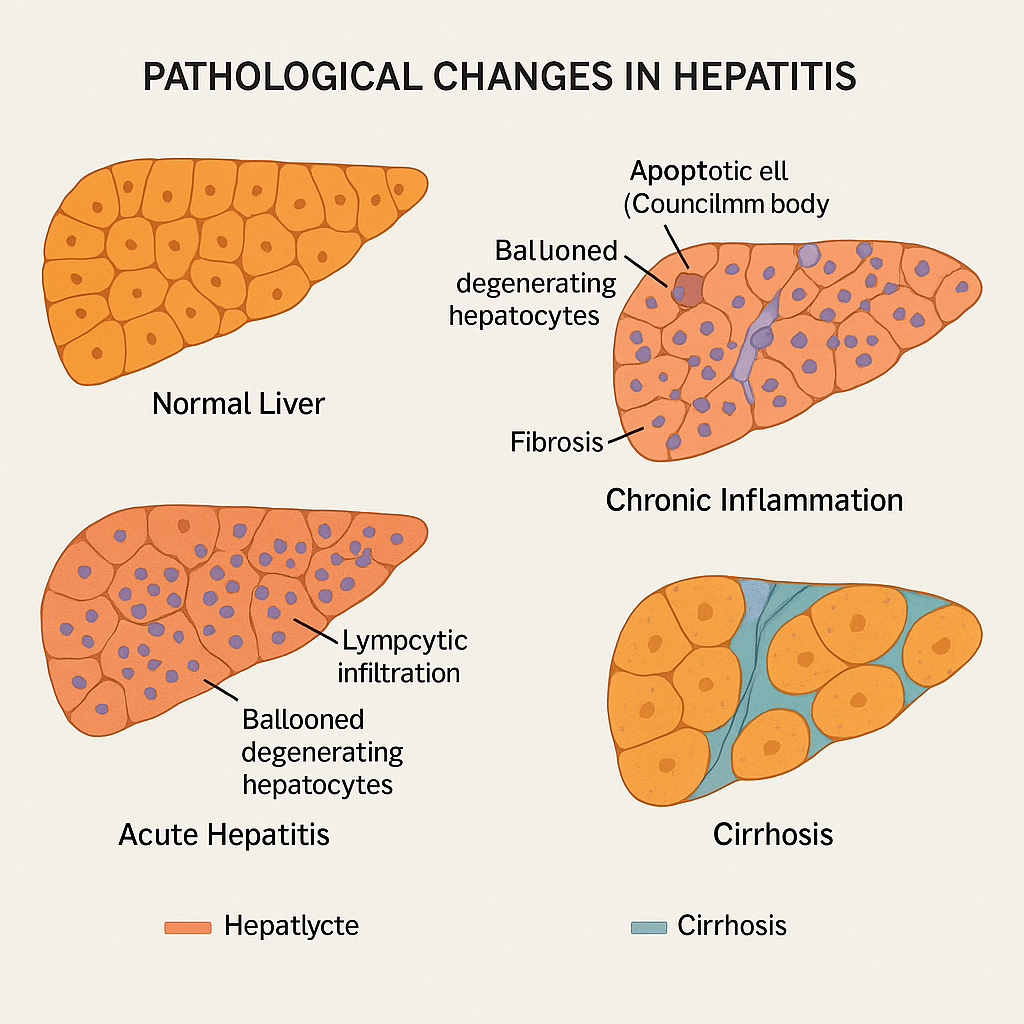
🧫 Pathological Changes in Amoebic Liver Abscess (ALA)
(Detailed academic explanation in a structured, clinical, and visually engaging narrative)
An Amoebic Liver Abscess (ALA) is a localized collection of necrotic hepatic tissue caused by Entamoeba histolytica, a protozoan parasite. It is the most frequent extraintestinal complication of amoebiasis and typically occurs when trophozoites spread hematogenously from the colon (via portal circulation) to the liver.
🔍 1. Pathogenesis and Mechanism of Injury
🔹 A. Intestinal Invasion
- Begins with ingestion of E. histolytica cysts through contaminated food or water.
- In the colon, trophozoites emerge, invade the mucosa, and cause flask-shaped ulcers.
🔹 B. Hepatic Spread
- From colon → portal vein → liver parenchyma
- The right lobe is most commonly involved due to direct portal blood flow
🔹 C. Necrosis and Abscess Formation
- Trophozoites secrete proteolytic enzymes (e.g., cysteine proteinases) that:
- Lyse hepatocytes
- Disrupt sinusoidal endothelial cells
- Leads to liquefactive necrosis, forming a single or multiple abscess cavities filled with:
- Anchovy sauce-like pus (reddish-brown, odorless, lacks neutrophils)
🔬 2. Microscopic Pathological Features
| Zone | Features |
|---|---|
| Central Zone | Liquefied necrotic tissue (no neutrophils), appears pinkish-brown and amorphous |
| Intermediate Zone | Trophozoites of E. histolytica, erythrophagocytosis seen (diagnostic) |
| Peripheral Zone | Viable liver tissue with inflammatory infiltration, congestion, and fibrosis |
👁️ 3. Gross (Macroscopic) Features
- Solitary, round abscess cavity (usually in right lobe)
- Cavity filled with chocolate-colored, anchovy paste-like material
- Well-circumscribed, may rupture into pleura, peritoneum, or pericardium if untreated
⚠️ 4. Complications
- Rupture into pleural, peritoneal, or pericardial cavity → empyema, peritonitis
- Secondary bacterial infection of abscess
- Chronic fibrosis or scarring of liver tissue
- Amoeboma formation (mass-like granulomatous reaction, rare)
🧠 Clinical Correlation
- Symptoms: Fever, right upper quadrant pain, tender hepatomegaly
- Diagnosis: Serology (anti-amoebic antibodies), ultrasound/CT, aspiration of abscess fluid
- Stool examination may not detect cysts/trophozoites in extraintestinal disease
📊 Summary Table of Pathological Changes in Amoebic Liver Abscess
| Stage/Zone | Key Pathological Features |
|---|---|
| Colon invasion | Flask-shaped ulcers, mucosal breach |
| Liver invasion | Hematogenous spread to right lobe |
| Central necrosis | Liquefied tissue, anchovy sauce appearance |
| Intermediate zone | Trophozoites with ingested RBCs |
| Peripheral zone | Inflammatory border with viable hepatocytes |
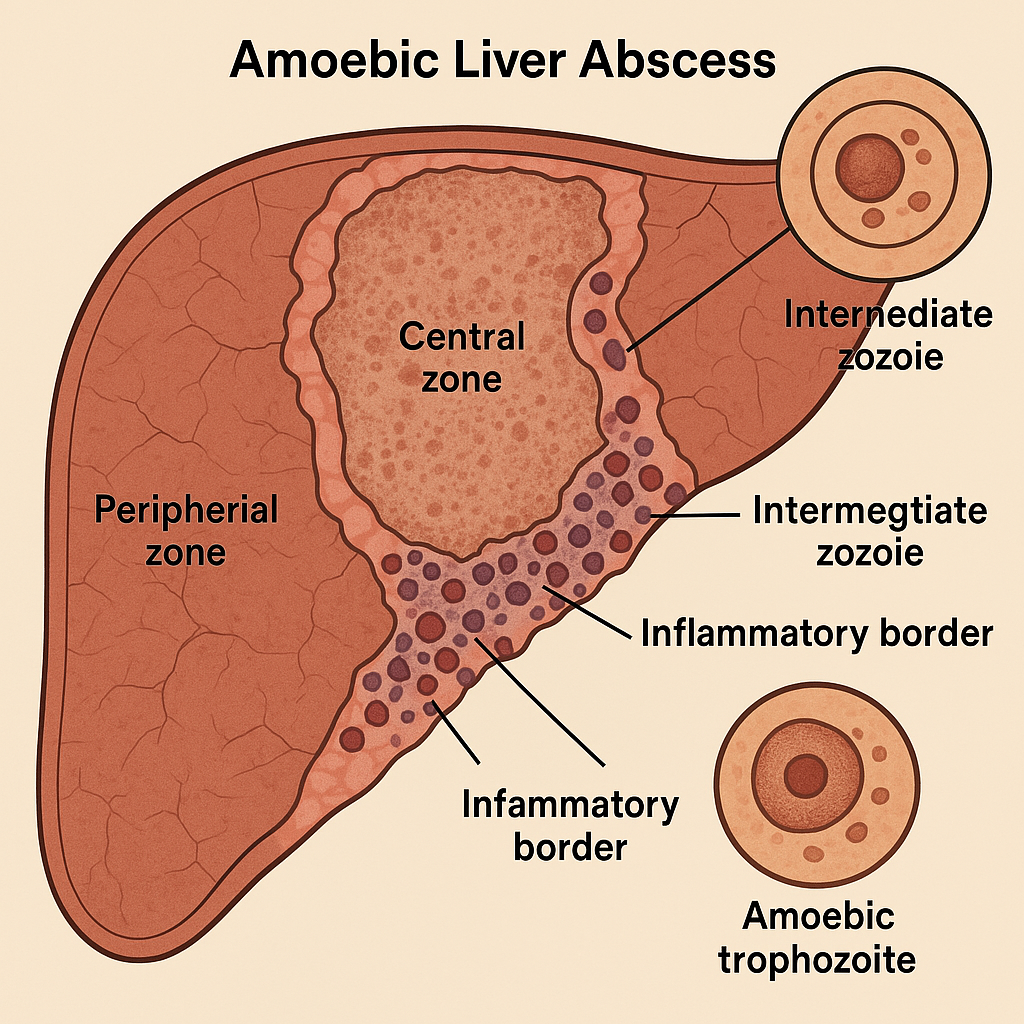
🧬 Pathological Changes in Cirrhosis of the Liver
Cirrhosis is a chronic progressive liver disease characterized by diffuse fibrosis, nodular regeneration of hepatocytes, and distortion of normal liver architecture. It is the final common pathway of many chronic liver injuries—whether due to alcohol, viral hepatitis, autoimmune disorders, metabolic diseases, or biliary obstructions.
Cirrhosis leads to portal hypertension, liver failure, and is a pre-neoplastic state for hepatocellular carcinoma (HCC).
🔍 1. Key Pathological Hallmarks
🔹 A. Bridging Fibrosis
- Fibrous septa connect portal tract to central vein, portal to portal, or central to central.
- Collagen deposition distorts vascular and parenchymal relationships.
🔹 B. Regenerative Nodules
- Surviving hepatocytes proliferate and form nodules surrounded by fibrosis.
- These nodules lack normal lobular organization (no central vein or portal triad inside).
- Nodules can be:
- Micronodular (<3 mm, uniform; common in alcoholic cirrhosis)
- Macronodular (>3 mm, variable size; often in post-hepatitic cirrhosis)
🔹 C. Distorted Liver Architecture
- Loss of normal sinusoidal pattern
- Abnormal vascular shunting
- Leads to functional decompensation and portal hypertension
🔬 2. Histopathological Changes
| Tissue Component | Changes in Cirrhosis |
|---|---|
| Hepatocytes | Atrophy, ballooning, fatty change, necrosis, and nodular regeneration |
| Sinusoids | Capillarization (loss of fenestrae), impaired blood flow |
| Stroma | Dense collagenous septa deposited by activated stellate (Ito) cells |
| Portal tracts | Blurred; difficult to identify due to fibrosis and inflammation |
🧫 3. Microscopic Features
- Fibrous septa cutting through parenchyma
- Regenerative nodules of hepatocytes
- Inflammatory infiltrate (especially in viral and autoimmune causes)
- Bile duct proliferation (especially in biliary cirrhosis)
- Loss of lobular architecture
👁️ 4. Gross (Macroscopic) Appearance
- Small, firm, nodular liver
- Surface is irregular with nodules separated by fibrous bands
- Color may vary:
- Yellow (fatty)
- Green (cholestatic)
- Brown (hemosiderin in hemochromatosis)
- Shrinkage of liver in late-stage disease
⚠️ 5. Complications of Cirrhosis
| Systemic Effect | Explanation |
|---|---|
| Portal Hypertension | Fibrosis impedes portal blood flow → splenomegaly, varices, ascites |
| Hepatic Encephalopathy | Ammonia and neurotoxin accumulation due to liver failure |
| Coagulopathy | Reduced synthesis of clotting factors |
| Hypoalbuminemia | Impaired protein synthesis |
| Hepatorenal Syndrome | Functional renal failure due to decreased perfusion |
| Hepatocellular Carcinoma (HCC) | Cirrhosis is a precancerous condition |
📊 Summary of Pathological Evolution
| Stage | Changes |
|---|---|
| Chronic liver injury | Inflammation, hepatocyte necrosis |
| Stellate cell activation | Collagen deposition and fibrosis |
| Bridging fibrosis | Loss of normal sinusoidal architecture |
| Regeneration | Formation of nodules |
| Cirrhosis | Distorted lobules, vascular shunts, portal hypertension |
🔬 Common Causes of Cirrhosis
- Alcoholic liver disease
- Chronic viral hepatitis B/C
- Non-alcoholic fatty liver disease (NAFLD/NASH)
- Autoimmune hepatitis
- Primary biliary cholangitis (PBC)
- Primary sclerosing cholangitis (PSC)
- Hemochromatosis, Wilson’s disease, Alpha-1-antitrypsin deficiency
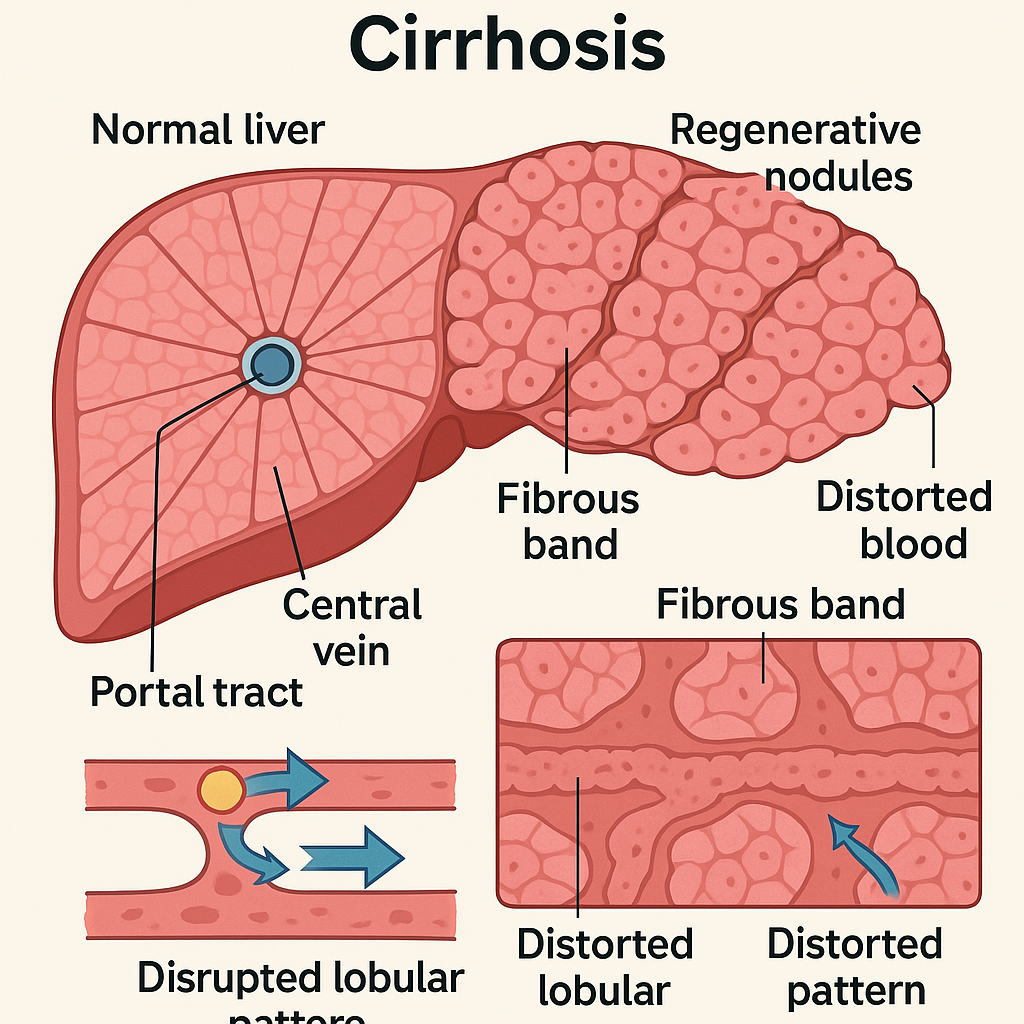
🌿 Cholecystitis – Pathological Changes
Cholecystitis is defined as the inflammation of the gallbladder, usually due to obstruction of the cystic duct, most commonly by gallstones (calculous cholecystitis). Less commonly, it may occur in the absence of stones (acalculous cholecystitis), especially in critically ill patients.
Let us walk through the sequence of pathological changes that occur in the gallbladder during cholecystitis.
🔬 1. Obstruction and Bile Stasis
- The initial event is typically obstruction of the cystic duct by a gallstone (or rarely, by a tumor, parasitic infestation, or stricture).
- This leads to bile stasis within the gallbladder, creating an ideal environment for bacterial overgrowth.
- Accumulated bile exerts chemical irritation on the mucosa and promotes inflammation.
🦠 2. Inflammation and Edema of the Wall
- Bacterial invasion (commonly E. coli, Klebsiella, Enterococcus) follows, leading to acute inflammation.
- Neutrophils infiltrate the mucosa and muscularis layer.
- The gallbladder wall becomes thickened and edematous due to:
- Vasodilation
- Increased vascular permeability
- Migration of leukocytes
🔥 3. Mucosal Damage and Ulceration
- Continued inflammation leads to mucosal ulceration, hemorrhage, and even mucosal sloughing.
- The inner lining may appear congested or necrotic in severe cases.
- Inflammatory exudate may contain pus, resulting in an empyema of the gallbladder.
💀 4. Necrosis and Gangrene (Severe Cases)
- If the inflammation progresses unchecked, the gallbladder wall may suffer ischemic necrosis.
- This is more likely in patients with:
- Atherosclerosis
- Sepsis
- Shock
- The gallbladder turns blackish-green and soft, indicating gangrenous cholecystitis.
🧨 5. Perforation and Complications
- As necrosis extends full-thickness, the wall may rupture, leading to:
- Biliary peritonitis
- Fistula formation
- Gallstone ileus (if a large stone enters the intestine)
- Abscess formation
🧱 Chronic Cholecystitis (Repeated Attacks)
- Long-standing inflammation results in:
- Fibrosis and scarring of the gallbladder wall
- Thickened, shrunken gallbladder
- Cholesterolosis (accumulation of cholesterol-laden macrophages)
- Porcelain gallbladder (calcification of the wall, predisposing to carcinoma)
- Lymphocytes and plasma cells predominate in chronic inflammation.
- Rokitansky-Aschoff sinuses (outpouchings of mucosa into the muscular wall) are often present.
🧠 Summary of Pathological Progression (Narrative Style)
- Initiation: Obstruction → bile retention.
- Acute response: Neutrophilic inflammation → wall thickening and mucosal damage.
- Suppuration: Empyema or pus accumulation in lumen.
- Necrosis: Wall ischemia → gangrenous transformation.
- Rupture: Full-thickness perforation with serious complications.
- Chronicity: Repeated inflammation → fibrosis, scarring, and calcification.
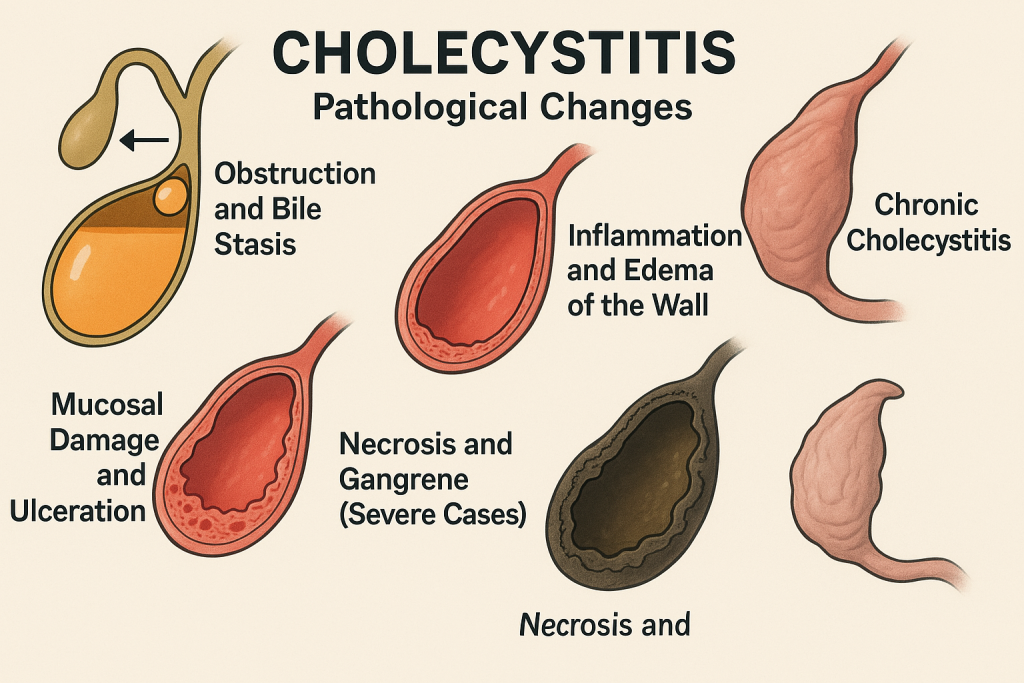
🔥 Pancreatitis – Pathological Changes
Pancreatitis refers to inflammation of the pancreas, an organ with both exocrine and endocrine functions. The pathology can be classified into two major forms:
- 🔹 Acute Pancreatitis
- 🔹 Chronic Pancreatitis
Each has distinct pathological features, but both involve the self-digestion of pancreatic tissue by its own enzymes.
⚡ I. Acute Pancreatitis
Acute pancreatitis is a sudden inflammatory reaction of the pancreas due to premature activation of digestive enzymes, especially trypsin, lipase, and elastase, within the pancreas itself.
🌀 Pathological Cascade:
1. Initiation by Acinar Cell Injury
- Triggered by alcohol, gallstones, trauma, infections, drugs, or metabolic disorders.
- These factors cause early activation of trypsinogen into trypsin within acinar cells.
2. Autodigestion of Pancreatic Tissue
- Activated trypsin activates other enzymes like lipase and elastase.
- Lipase → Fat necrosis by breaking down fat into fatty acids.
- Elastase → Digests elastic tissue in blood vessels, causing hemorrhage.
3. Acute Inflammation
- Massive neutrophilic infiltration.
- Edema and interstitial fluid accumulation due to increased vascular permeability.
- Cytokines and inflammatory mediators (e.g., TNF-α, IL-6) promote systemic inflammation.
4. Tissue Necrosis
- Parenchymal cells undergo coagulative necrosis.
- Peripancreatic fat undergoes fat necrosis (chalky white calcium soaps – saponification).
- May result in hemorrhagic pancreatitis—a severe, life-threatening variant.
5. Complications of Acute Pancreatitis
- Pancreatic pseudocyst formation (walled-off necrosis)
- Acute fluid collections or abscesses
- Systemic inflammatory response syndrome (SIRS) and multi-organ failure
🌲 II. Chronic Pancreatitis
Chronic pancreatitis is a progressive, irreversible inflammation leading to permanent structural damage and functional loss of the pancreas.
🌀 Pathological Changes Over Time:
1. Repeated Inflammation
- Most commonly due to alcohol abuse, autoimmune disease, or hereditary factors.
- Persistent inflammation causes fibrosis and atrophy of acinar cells.
2. Loss of Exocrine Tissue
- Acinar cells degenerate and are replaced by fibrous tissue.
- Leads to malabsorption, steatorrhea, and weight loss.
3. Ductal Changes
- Obstruction and dilation of pancreatic ducts due to:
- Protein plugs
- Fibrotic strictures
- Intraluminal calcification → pancreatic stones
4. Islet Cell Destruction
- In later stages, endocrine (islet) cells are also destroyed.
- May result in secondary diabetes mellitus (pancreatogenic diabetes).
5. Chronic Fibrosis and Atrophy
- The pancreas becomes shrunken, fibrotic, and calcified.
- Ducts become dilated and irregular.
- Inflammatory infiltrate is mainly lymphocytes and plasma cells (unlike neutrophils in acute).
🧠 Summary Comparison of Pathological Changes
| Feature | Acute Pancreatitis | Chronic Pancreatitis |
|---|---|---|
| Onset | Sudden, rapid | Slow, insidious |
| Main process | Autodigestion + acute inflammation | Fibrosis + irreversible damage |
| Enzymatic action | Trypsin, lipase, elastase → tissue necrosis | Minimal enzymatic activity in later stages |
| Inflammatory cells | Neutrophils | Lymphocytes, plasma cells |
| Complications | Pseudocysts, hemorrhage, ARDS | Diabetes, steatorrhea, pancreatic stones |
| Outcome | Potentially reversible | Irreversible, progressive |
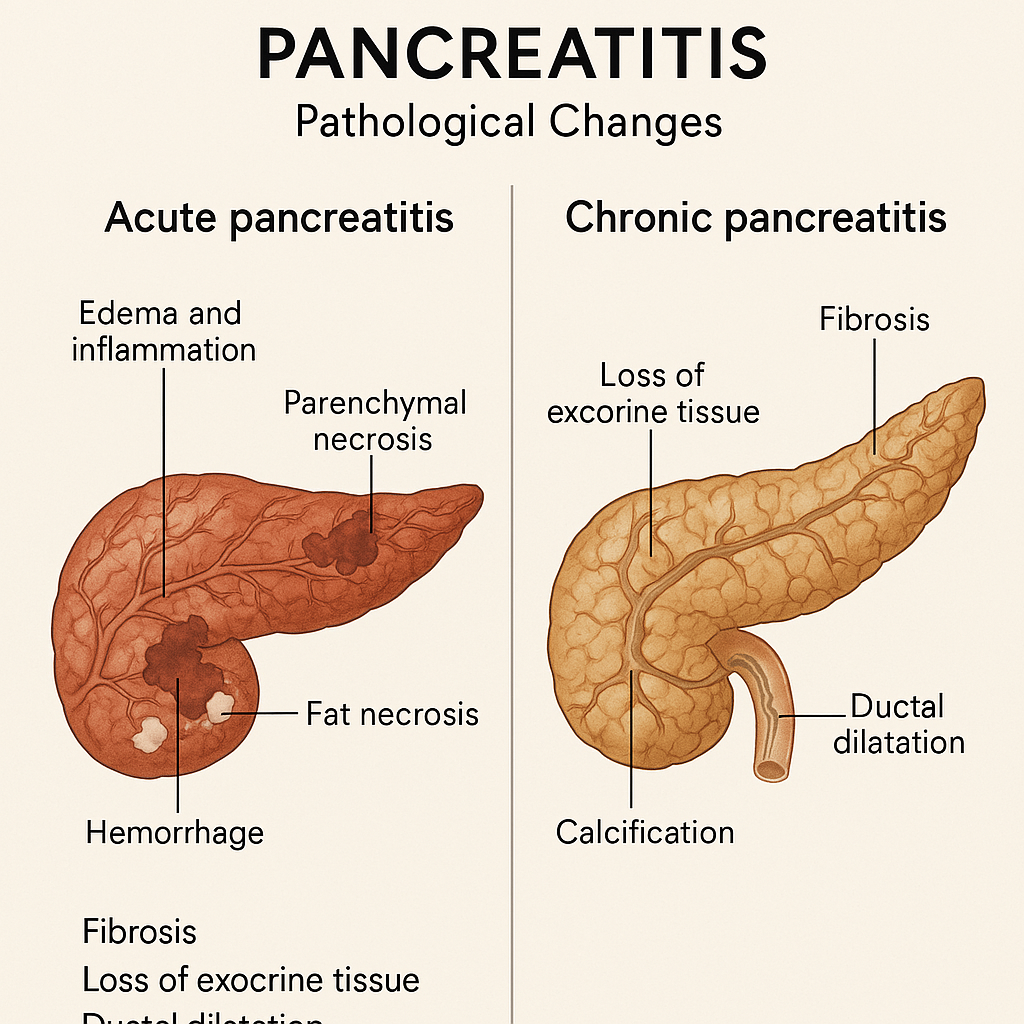
🧬 Pathological Changes in Liver Tumors
Tumors of the liver may arise primarily within the liver (primary tumors) or result from the spread of cancers from other organs (secondary/metastatic tumors). Liver tumors can be benign or malignant, with the latter being far more common and clinically significant.
🔵 I. Benign Liver Tumors – Pathological Changes
1. Hepatic Hemangioma
- Most common benign tumor of the liver.
- Composed of vascular channels lined by endothelium and supported by fibrous tissue.
- Grossly appears as a well-circumscribed, reddish-blue lesion.
- Usually non-invasive and asymptomatic, but large hemangiomas may bleed.
2. Hepatocellular Adenoma
- Arises from hepatocytes, often in women using oral contraceptives.
- Well-encapsulated tumor with a soft, tan-yellow cut surface.
- Histologically shows normal hepatocytes without portal tracts or bile ducts.
- Can undergo necrosis or hemorrhage, especially during pregnancy or trauma.
- Rare risk of transformation to hepatocellular carcinoma (HCC).
🔴 II. Malignant Liver Tumors – Pathological Changes
Malignant tumors of the liver may be:
- Primary: Originating within liver tissue
- Secondary (metastatic): Spread from colon, lung, breast, stomach, pancreas, etc.
🔥 A. Hepatocellular Carcinoma (HCC)
The most common primary liver cancer, often linked to chronic liver disease (e.g., Hepatitis B/C, cirrhosis, aflatoxins).
🔬 Pathological Changes:
🟠 1. Initiation by Chronic Injury
- Chronic hepatitis or alcohol abuse leads to repeated hepatocyte injury.
- Cellular regeneration + inflammation → DNA damage and mutation.
🟠 2. Dysplasia
- Hepatocytes undergo atypical hyperplasia (dysplasia), progressing from:
- Low-grade → High-grade dysplasia → Carcinoma in situ
🟠 3. Tumor Nodules Formation
- Transforms into solid tumor nodules, which may:
- Be single or multiple
- Vary in color: yellow (lipid), greenish (bile), or hemorrhagic
🟠 4. Histological Hallmarks
- Trabecular, pseudoacinar, or solid patterns of hepatocytes
- Hyperchromatic nuclei, mitoses, loss of normal architecture
- Tumor cells may produce bile or alpha-fetoprotein (AFP)
🟠 5. Invasion and Spread
- Tumor invades portal and hepatic veins → leads to vascular invasion
- Commonly metastasizes to:
- Lungs
- Bones
- Peritoneum
🟣 B. Cholangiocarcinoma (Bile Duct Cancer)
Adenocarcinoma arising from intrahepatic or extrahepatic bile ducts.
🧬 Pathological Features:
- Dense fibrotic stroma (desmoplastic reaction)
- Glandular structures formed by malignant epithelial cells
- Tumors often appear as white, firm masses, unlike soft HCC
🟠 C. Angiosarcoma of Liver
- Rare, aggressive tumor derived from endothelial cells.
- Associated with exposure to vinyl chloride, arsenic, Thorotrast.
- Shows vascular channels, pleomorphic endothelial cells, and hemorrhagic areas.
- High rate of metastasis and fatality.
🔺 D. Metastatic Tumors of the Liver
- The most common type of liver malignancy overall.
- Liver is a frequent site of metastasis due to its rich dual blood supply (portal vein + hepatic artery).
- Common primary sites: colon, breast, lung, stomach, pancreas.
Pathological Appearance:
- Multiple well-demarcated nodules scattered across the liver.
- Central necrosis may produce “umbilicated” appearance.
- Histology mimics the primary tumor (e.g., glandular for colon, squamous for lung, etc.)
🧠 Summary of Key Pathological Changes
| Tumor Type | Origin | Key Pathology |
|---|---|---|
| Hemangioma | Blood vessels | Cavernous vascular spaces, no atypia |
| Hepatocellular Adenoma | Hepatocytes | Normal hepatocytes, no bile ducts, risk of hemorrhage |
| HCC | Hepatocytes | Dysplasia → carcinoma, bile production, vascular invasion |
| Cholangiocarcinoma | Bile duct epithelium | Adenocarcinoma with dense fibrosis, glandular formation |
| Angiosarcoma | Endothelium | Highly vascular, hemorrhagic, aggressive |
| Metastases | Extrahepatic origin | Multiple nodules mimicking primary tumor |
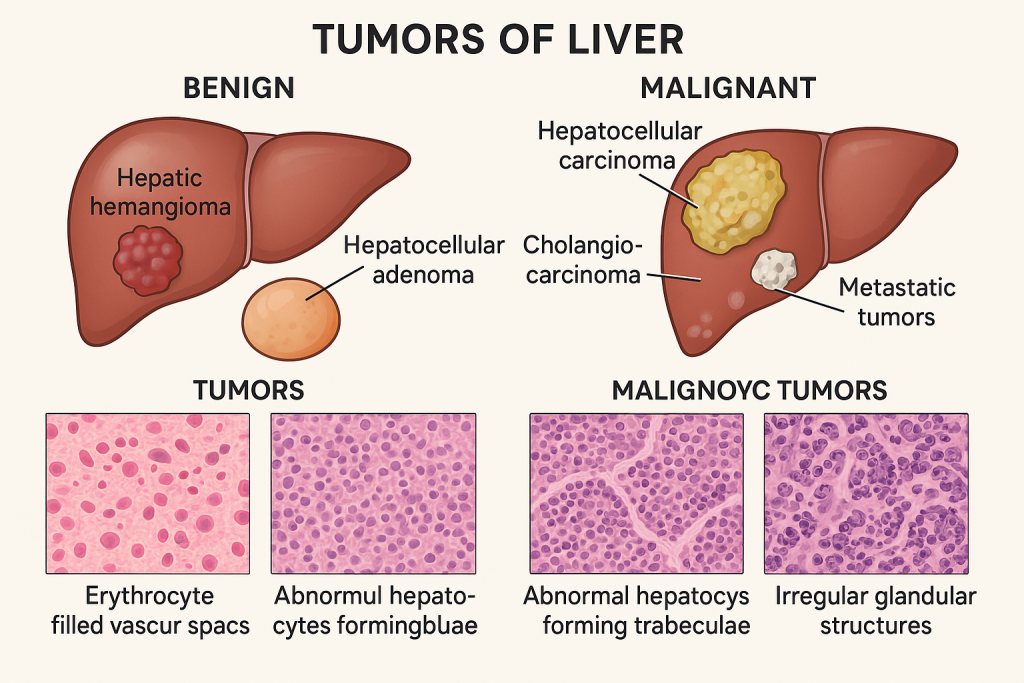
🧬 Pathological Changes in Gallbladder Tumors
Gallbladder tumors are relatively rare but highly aggressive, with most being malignant. They often present late due to nonspecific symptoms, and by the time of diagnosis, they frequently invade locally or metastasize.
Tumors may be classified as:
- 🔹 Benign (rare)
- 🔴 Malignant (common – mostly adenocarcinomas)
🔵 I. Benign Tumors of the Gallbladder
Benign tumors are uncommon and include:
- Adenomas (true epithelial neoplasms)
- Cholesterol polyps (non-neoplastic)
- Leiomyomas, fibromas (mesenchymal origin)
🔍 Pathological Features of Gallbladder Adenoma:
- Usually small (<2 cm), sessile or pedunculated growths.
- Composed of well-differentiated glandular epithelium.
- No evidence of invasion or metastasis.
- May coexist with cholelithiasis and chronic inflammation.
- Rare potential for malignant transformation.
🔴 II. Malignant Tumors of the Gallbladder
The majority of gallbladder malignancies are:
- Adenocarcinomas (95%)
- Less commonly: squamous cell carcinoma, adenosquamous, small-cell carcinoma, sarcoma
🔬 Pathological Changes in Gallbladder Adenocarcinoma
🧠 1. Initiation by Chronic Irritation
- Long-standing cholelithiasis (gallstones) is the most common risk factor.
- Chronic inflammation and epithelial injury lead to intestinal metaplasia and dysplasia.
🔁 Sequence:
Normal mucosa → Chronic cholecystitis → Metaplasia → Dysplasia → Carcinoma in situ → Invasive carcinoma
🧬 2. Dysplasia to Invasion
- Dysplastic epithelial cells exhibit:
- Enlarged nuclei, hyperchromasia
- Loss of polarity and increased mitoses
- Carcinoma in situ remains confined to mucosa.
- With time, malignant cells breach the basement membrane and invade deeper layers:
- Lamina propria
- Muscularis
- Perimuscular connective tissue
🌡️ 3. Desmoplastic Reaction & Local Spread
- Tumor induces desmoplasia (fibrotic response).
- Infiltrates into:
- Liver parenchyma
- Common bile duct
- Duodenum or transverse colon
🧫 4. Histopathological Subtypes
- Infiltrative type: poorly defined thickening of gallbladder wall.
- Papillary type: exophytic, friable growth with better prognosis.
Histologically, the tumor shows:
- Irregular glands lined by atypical cuboidal to columnar cells
- High nuclear/cytoplasmic ratio
- Mucin production in many cases
- Invasion of perineural spaces and blood vessels
⚠️ 5. Metastasis
- Early spread due to lack of serosa in gallbladder.
- Regional lymph nodes: cystic, periportal, coeliac
- Distant metastases: liver, lungs, peritoneum
🧠 Summary of Pathological Evolution
| Stage | Key Changes |
|---|---|
| Normal Mucosa | Simple columnar epithelium |
| Chronic Cholecystitis | Inflammatory infiltrate, mucosal damage |
| Intestinal Metaplasia | Replacement with goblet cells |
| Dysplasia | Cellular atypia, loss of normal structure |
| Carcinoma in situ | Confined neoplastic cells |
| Invasive Adenocarcinoma | Infiltration into muscular wall and beyond |
| Metastatic Spread | Lymphatic, hematogenous, direct invasion |
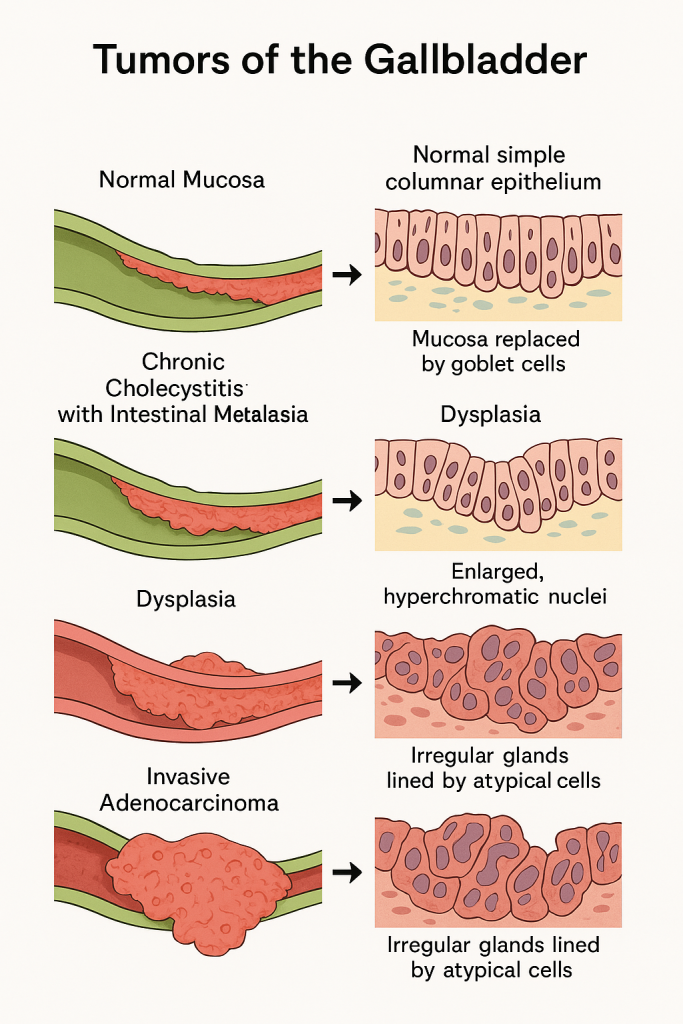
🧬 Pathological Changes in Tumors of the Pancreas
Tumors of the pancreas may be benign or malignant, and they arise from either:
- The exocrine pancreas (ductal and acinar cells)
- Or the endocrine pancreas (islets of Langerhans)
Broad classification:
🔹 Exocrine tumors – most common and usually malignant
🔹 Endocrine tumors (Pancreatic Neuroendocrine Tumors, PNETs) – rare, may be functional or non-functional
🔴 I. Exocrine Tumors (Mainly Malignant)
A. Pancreatic Ductal Adenocarcinoma (PDAC)
This is the most common and most lethal tumor of the pancreas, typically arising from the ductal epithelium and often located in the head of the pancreas.
🔬 Pathological Changes:
🧠 1. Precancerous Lesions (PanIN)
- Begins with Pancreatic Intraepithelial Neoplasia (PanIN).
- Progression:
- PanIN-1 → Low-grade dysplasia (mild atypia)
- PanIN-2 → Moderate dysplasia
- PanIN-3 → Carcinoma in situ (severe dysplasia)
🔄 2. Malignant Transformation
- Cells exhibit:
- Nuclear enlargement, pleomorphism
- Loss of polarity, increased mitotic activity
- Basement membrane is breached, leading to invasion into the stroma.
🔥 3. Infiltrative Growth
- Dense desmoplastic stroma is a hallmark of PDAC.
- Infiltrates peripancreatic fat, duodenum, spleen, and mesentery.
🔬 4. Histopathology
- Irregular ductal/glandular structures lined by atypical columnar cells.
- Perineural and lymphovascular invasion is common.
- Tumor induces fibrosis and may compress surrounding structures (like the common bile duct).
⚠️ 5. Metastasis
- Common sites: liver, peritoneum, lungs, bones.
- Early spread via lymphatics and blood vessels.
🟠 II. Cystic Tumors of the Pancreas
Include both benign and malignant varieties:
A. Serous Cystadenoma (Benign)
- Composed of glycogen-rich cuboidal cells
- Forms a honeycomb-like cystic structure
B. Mucinous Cystic Neoplasm (MCN) (Premalignant to malignant)
- Lined by mucin-producing columnar epithelium
- Often contains ovarian-type stroma
- Risk of progression to mucinous cystadenocarcinoma
C. Intraductal Papillary Mucinous Neoplasm (IPMN)
- Affects main or branch pancreatic ducts
- Papillary projections into ducts, with mucin overproduction
- Can progress to invasive adenocarcinoma
🟣 III. Endocrine Tumors (Pancreatic Neuroendocrine Tumors – PNETs)
These tumors arise from islet cells and may be:
- Functional (producing hormones like insulin, glucagon, gastrin)
- Non-functional (often malignant and aggressive)
🔬 Pathological Features:
- Tumor cells arranged in nests, trabeculae, or rosettes
- Uniform round nuclei, salt-and-pepper chromatin
- May express chromogranin A and synaptophysin
- Well-differentiated PNETs can be benign or low-grade malignant
- Poorly differentiated neuroendocrine carcinoma is highly malignant
🧠 Summary Table – Pathological Progression
| Tumor Type | Origin | Key Pathological Features |
|---|---|---|
| PDAC | Ductal epithelium | Glandular pattern, dense stroma, invasion, metastasis |
| Serous cystadenoma | Acinar cells | Small cysts lined by cuboidal epithelium, benign |
| MCN | Mucinous epithelial cells | Mucin production, ovarian-type stroma, malignant potential |
| IPMN | Ductal epithelium | Papillary growth in ducts, mucinous, variable dysplasia |
| PNET | Islet cells | Neuroendocrine markers, trabecular pattern, variable malignancy |
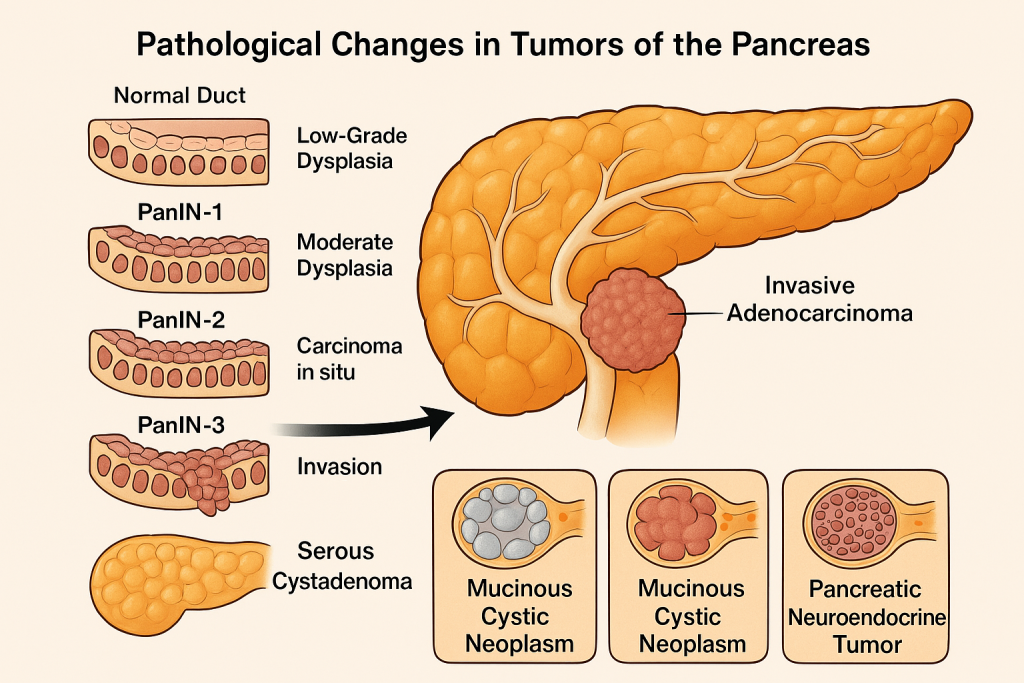
Skeletal system
🦴 Bone Healing – Pathological Changes
Bone healing is a highly organized biological process that restores the structural integrity and function of fractured or surgically cut bone. Unlike other tissues, bone heals without forming a scar, by regenerating new bone tissue.
It occurs in three overlapping phases:
🔹 1. Inflammatory Phase (Reactive Phase)
🔹 2. Reparative Phase
🔹 3. Remodeling Phase (Maturation Phase)
🔥 1. Inflammatory Phase (Day 0 to Day 7)
This is the immediate response to fracture or bone injury.
🧬 Pathological Changes:
- Hematoma formation at the fracture site due to torn blood vessels.
- Release of platelet-derived growth factors (PDGF) and cytokines.
- Fibrin clot acts as a scaffold.
- Inflammatory cells (neutrophils, macrophages) infiltrate the area.
- Removal of necrotic debris and bone fragments (via phagocytosis).
🧠 Purpose: Initiates repair by preparing the site for new tissue formation.
🌱 2. Reparative Phase (Day 7 to Week 6)
This is the active tissue regeneration phase.
🧬 Pathological Changes:
🟡 A. Soft Callus Formation (Fibrocartilaginous Callus)
- Fibroblasts and chondroblasts infiltrate hematoma.
- Formation of granulation tissue rich in collagen and cartilage.
- Stabilizes the fracture but does not yet bear weight.
- Appears radiolucent (less dense) on X-ray.
🟠 B. Hard Callus Formation (Woven Bone)
- Osteoblasts start laying down woven bone over the soft callus.
- New bone bridges the fracture gap.
- Gradually mineralizes (calcium and phosphate deposition).
- Appears more radio-opaque on X-ray.
🧠 Purpose: Restores mechanical strength to the bone.
🔁 3. Remodeling Phase (Months to Years)
This is the final stage, where immature bone is replaced by mature lamellar bone.
🧬 Pathological Changes:
- Woven bone is resorbed by osteoclasts.
- Replaced by organized lamellar bone with correct architecture.
- Haversian systems and medullary canal are restored.
- Bone regains normal strength and function.
- The callus becomes less prominent or disappears on X-ray.
🧠 Purpose: Restores bone’s shape, structure, and biomechanical function.
🔎 Additional Factors Influencing Pathological Healing
- 🚭 Smoking → reduces vascularity and oxygenation
- 🍽️ Malnutrition → reduces collagen and bone matrix formation
- 🦠 Infection → causes osteomyelitis and delays healing
- 💉 Medications → NSAIDs and corticosteroids may impair healing
- 🧬 Age, diabetes, and osteoporosis also slow bone regeneration
🧠 Summary Flowchart of Bone Healing
| Phase | Key Events |
|---|---|
| Inflammatory | Hematoma, cytokines, inflammation, debris removal |
| Reparative | Granulation tissue → soft callus → woven bone (hard callus) |
| Remodeling | Resorption of woven bone → lamellar bone formation → restoration of function |
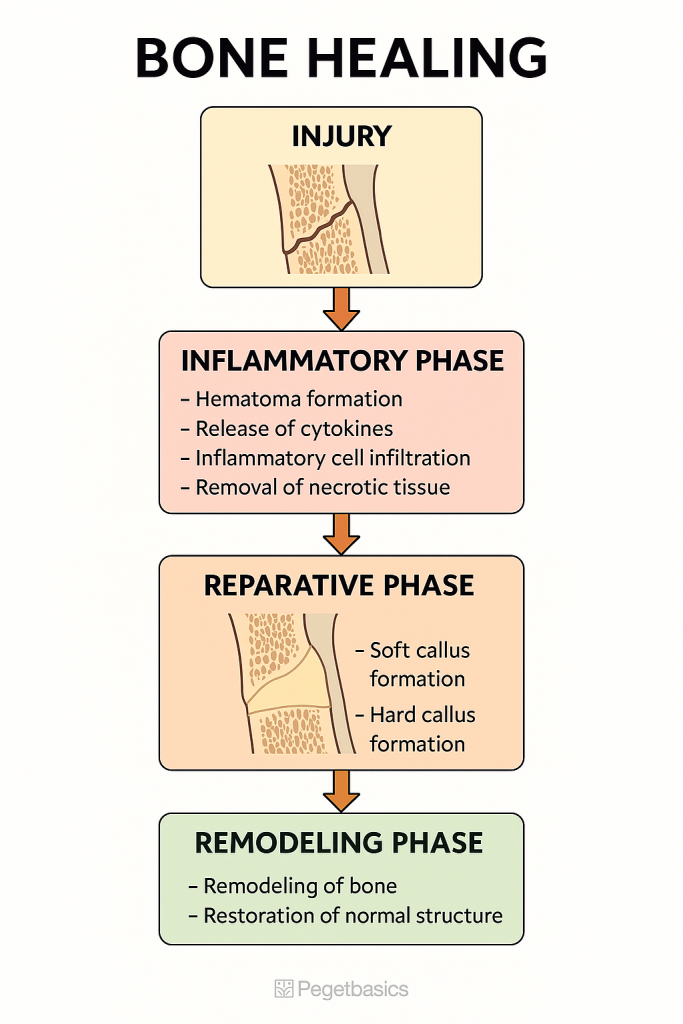
🦴 Osteoporosis – Pathological Changes
Osteoporosis is a systemic skeletal disorder characterized by:
- Reduction in total bone mass
- Disruption of bone microarchitecture
- Increased bone fragility, leading to a higher risk of fractures
Despite the loss of quantity, the quality of bone mineralization remains normal. This distinguishes osteoporosis from osteomalacia, where bone mineralization is defective.
🔬 Pathogenesis: Key Pathological Changes
Osteoporosis results from an imbalance in bone remodeling — the ongoing process by which old bone is resorbed by osteoclasts and new bone is formed by osteoblasts.
🔹 1. Increased Bone Resorption
- Triggered by hormonal changes (especially estrogen deficiency in postmenopausal women), aging, physical inactivity, or calcium/vitamin D deficiency.
- Osteoclasts become more active, leading to faster bone breakdown.
🔹 2. Decreased Bone Formation
- Osteoblast activity declines, especially with aging.
- Fewer new bone matrix proteins (like collagen) are synthesized.
- Impaired osteoblast differentiation and lifespan.
🔹 3. Trabecular Bone Loss
- Trabeculae become thin, perforated, or completely lost.
- Reduced connectivity leads to a weakened internal framework.
- Bone marrow spaces appear widened on histology.
🔹 4. Cortical Bone Thinning
- Cortical (compact) bone also thins and becomes porous.
- Enlarged Haversian and Volkmann’s canals.
- Bone shaft diameter increases, but overall strength declines.
🔹 5. Microfractures and Mechanical Failure
- Cumulative structural compromise causes:
- Microcracks
- Reduced ability to absorb mechanical energy
- Easy fracture under normal stress
🧪 Histological Features
| Normal Bone | Osteoporotic Bone |
|---|---|
| Dense trabeculae, good connectivity | Thin, sparse trabeculae |
| Balanced bone turnover | Increased osteoclasts, fewer osteoblasts |
| Well-mineralized matrix | Still mineralized but reduced in quantity |
| Compact cortical bone | Porous, thinned cortex |
🧠 Key Clinical Consequences of Pathological Changes
- Fragility fractures from minor trauma
- Vertebral compression → kyphosis, loss of height
- Femoral neck fractures → immobility, morbidity
- Wrist fractures → common in postmenopausal women
- Chronic pain and reduced mobility
- Decreased quality of life
🔄 Summary of Pathological Progression
🔸 Normal bone remodeling
⬇
🔸 Estrogen deficiency / Aging / Inactivity
⬇
🔸 ↑ Bone resorption (osteoclasts) + ↓ bone formation (osteoblasts)
⬇
🔸 Trabecular thinning, cortical porosity
⬇
🔸 Microarchitectural weakness
⬇
🔸 Fractures and skeletal deformities
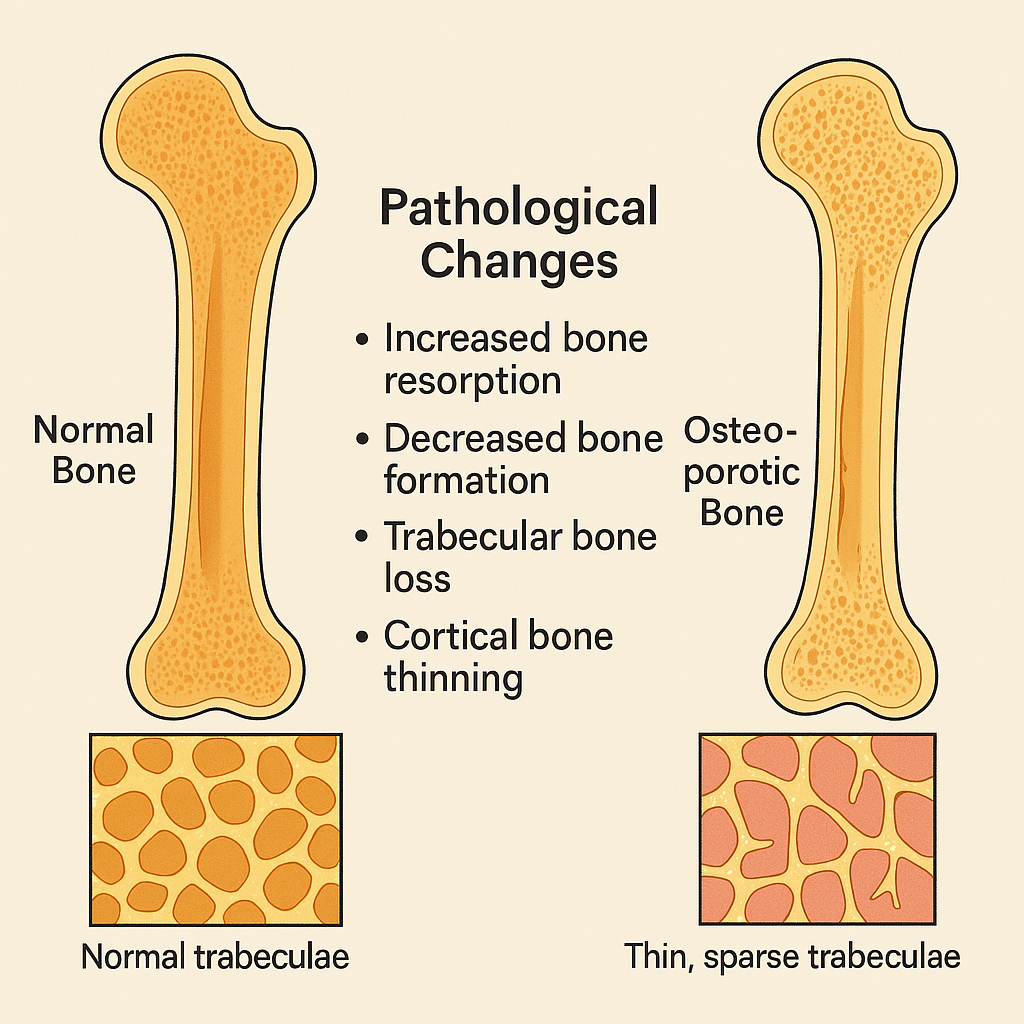
🦴 Osteomyelitis – Pathological Changes
Osteomyelitis is an infection of the bone and bone marrow, typically caused by bacteria, though fungi and mycobacteria may also be responsible. The pathological process varies depending on whether the infection is acute, subacute, or chronic.
The most common causative organism is Staphylococcus aureus, which gains entry to bone through:
- Hematogenous spread
- Direct inoculation (e.g., trauma, surgery)
- Contiguous spread (e.g., diabetic ulcers)
🔥 I. Acute Osteomyelitis – Pathological Changes
🧬 1. Initial Bacterial Invasion
- Bacteria lodge in the metaphysis of long bones (especially in children due to rich blood supply and slow circulation).
- The infection triggers an acute inflammatory response.
🔥 2. Suppurative Inflammation
- Neutrophils infiltrate the marrow spaces.
- Pus forms and increases intramedullary pressure.
- The exudate spreads through Volkmann’s and Haversian canals into the periosteum.
💀 3. Vascular Compromise and Bone Necrosis
- Rising pressure compresses blood vessels → ischemia and bone death.
- This leads to formation of a sequestrum:
- A fragment of necrotic bone that is separated from viable bone.
- Nonviable, avascular, and often surrounded by pus.
🧱 4. Periosteal Reaction
- The periosteum lifts, stimulating new bone formation by osteoblasts.
- A layer of reactive new bone (called involucrum) forms around the sequestrum.
🔁 Cycle: Infection → Necrosis (Sequestrum) → New Bone (Involucrum) → Chronic inflammation
🌿 II. Chronic Osteomyelitis – Pathological Changes
Occurs when acute infection is inadequately treated, or the host immune response is impaired.
🦠 1. Persistent Infection
- Pockets of infection remain walled off inside bone.
- Continuous or intermittent pus drainage through sinus tracts to the skin.
🧬 2. Granulation and Fibrosis
- Chronic inflammation leads to formation of granulation tissue.
- This tissue replaces normal marrow and contributes to fibrosis and sclerosis of surrounding bone.
🧫 3. Histological Changes
- Chronic inflammatory cells: lymphocytes, plasma cells, macrophages.
- Sequestra surrounded by inflammatory exudates.
- Involucrum with irregular new bone trabeculae.
- Sinus tracts lined by granulomatous epithelium.
- Sometimes, Brodie’s abscess: localized abscess in bone surrounded by sclerotic wall.
🧠 Summary of Pathological Progression
| Stage | Pathological Changes |
|---|---|
| Acute Stage | Neutrophilic infiltration, marrow edema, pus formation, ischemic necrosis |
| Sequestrum | Necrotic, devitalized bone separated from living bone |
| Involucrum | Reactive new bone formation around the necrotic area |
| Chronic Stage | Granulation tissue, fibrosis, sinus tracts, persistent infection |
| Complications | Chronic draining sinuses, squamous cell carcinoma, amyloidosis |
📍 Key Clinical Correlates
- Pain, fever, swelling, localized tenderness
- Elevated ESR and CRP, leukocytosis
- Radiologic findings: lytic lesions, periosteal elevation, sequestra
- Definitive diagnosis: bone biopsy and culture
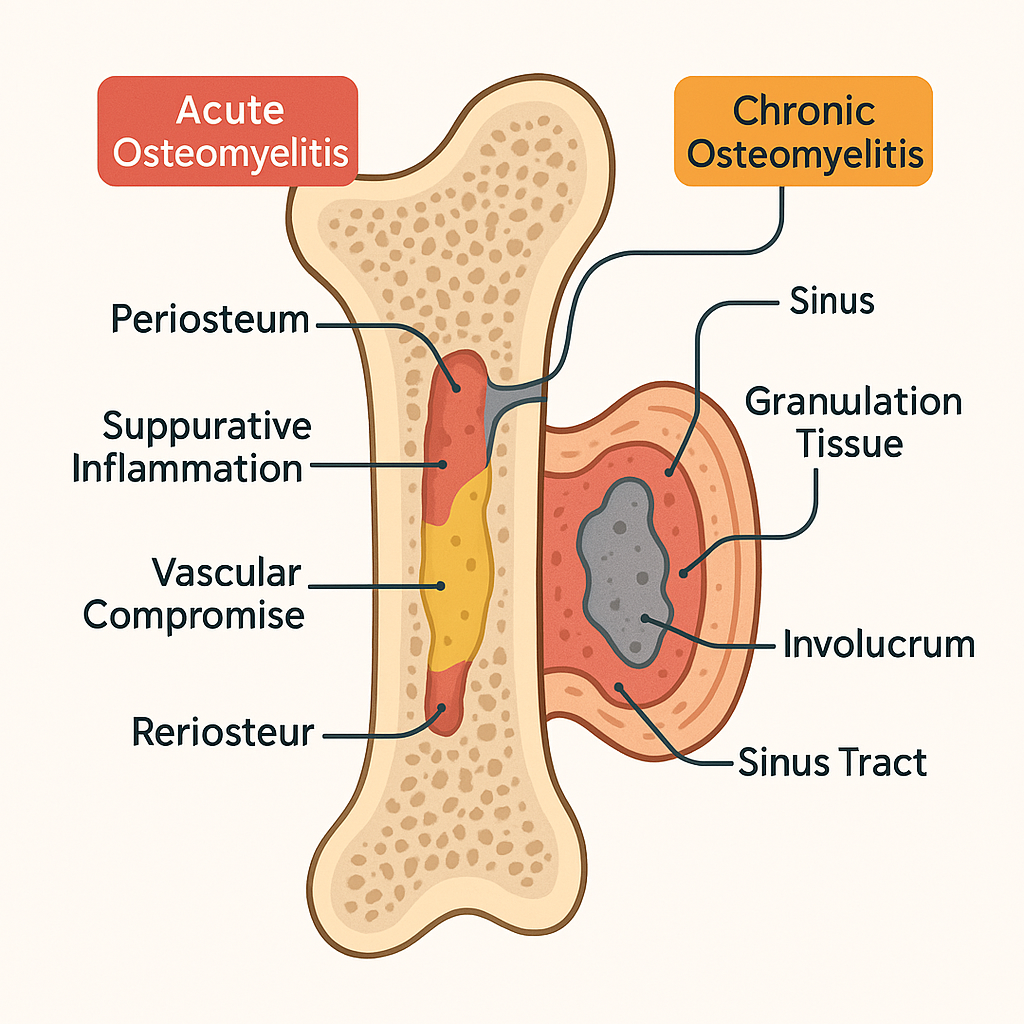
🦴 Bone Tumors – Pathological Changes
Bone tumors are abnormal growths that arise from bone tissue and may be:
- 🔹 Benign: Localized, slow-growing, well-differentiated, non-invasive
- 🔴 Malignant: Rapidly growing, poorly differentiated, invasive, and capable of metastasis
Tumors can arise from any bone-forming or supporting element, including:
- Osteoblasts (bone matrix-forming)
- Chondrocytes (cartilage-forming)
- Fibroblasts
- Bone marrow components
🔵 I. Benign Bone Tumors – Pathological Changes
1. Osteoid Osteoma
- Small (<2 cm), usually in cortex of long bones.
- Composed of woven bone trabeculae surrounded by osteoblasts and vascularized stroma.
- Surrounded by sclerotic bone due to reactive changes.
- Central “nidus” is the hallmark.
2. Osteochondroma
- Bony exostosis with a cartilage cap, usually near growth plates.
- The lesion grows outward from the bone surface.
- Histology shows hyaline cartilage cap with underlying trabecular bone.
- Benign but can rarely transform into chondrosarcoma.
3. Enchondroma
- Arises from medullary cavity.
- Composed of benign hyaline cartilage nodules, often calcified.
- May expand and thin the overlying cortex.
🔴 II. Malignant Bone Tumors – Pathological Changes
1. Osteosarcoma (Most common primary malignant bone tumor)
- Affects metaphysis of long bones (esp. around knee).
- Malignant osteoblasts produce osteoid matrix.
- Histology:
- Highly pleomorphic cells
- Hyperchromatic nuclei, mitotic figures
- Areas of necrosis and hemorrhage
- Irregular osteoid deposition in lace-like pattern
- Infiltrates surrounding soft tissues and may metastasize to lungs.
2. Chondrosarcoma
- Malignant cartilage-forming tumor, often in pelvis or ribs.
- Gross: Lobulated, bluish-white tumor with gelatinous consistency.
- Histology:
- Atypical chondrocytes in lacunae
- Multinucleated cells, binucleation
- Myoid background with calcification
- Invasion into bone trabeculae
3. Ewing Sarcoma
- Highly aggressive, in diaphysis of long bones, mostly in children/adolescents.
- Arises from primitive neuroectodermal cells.
- Histology:
- Sheets of small round blue cells
- Scant cytoplasm, round nuclei
- Homer Wright rosettes may be seen
- Associated with t(11;22) translocation
4. Giant Cell Tumor (Osteoclastoma)
- Typically occurs at the epiphysis of long bones.
- Composed of:
- Mononuclear stromal cells (neoplastic)
- Numerous multinucleated giant cells (osteoclast-like)
- Can be locally aggressive and recur.
🧠 Pathological Hallmarks of Bone Tumors
| Tumor Type | Cells Involved | Matrix Produced | Key Features |
|---|---|---|---|
| Osteosarcoma | Osteoblasts | Osteoid | Pleomorphism, atypical mitoses, tumor osteoid |
| Chondrosarcoma | Chondrocytes | Cartilage | Binucleation, myxoid stroma, bone invasion |
| Ewing Sarcoma | Primitive cells | None | Round blue cells, rosettes, aggressive behavior |
| Osteoid Osteoma | Osteoblasts | Osteoid | Nidus of woven bone, reactive sclerosis |
| Giant Cell Tumor | Stromal + giant cells | None | Epiphyseal, osteoclast-like cells |
📍 Summary of Pathological Changes
- Benign Tumors: Well-defined, non-invasive, with slow proliferation of differentiated cells. Reactive bone formation may occur.
- Malignant Tumors: Cellular atypia, aggressive invasion, destruction of normal bone architecture, production of abnormal matrix (osteoid or cartilage).
- Vascular invasion, necrosis, and soft tissue extension are common in malignancies.
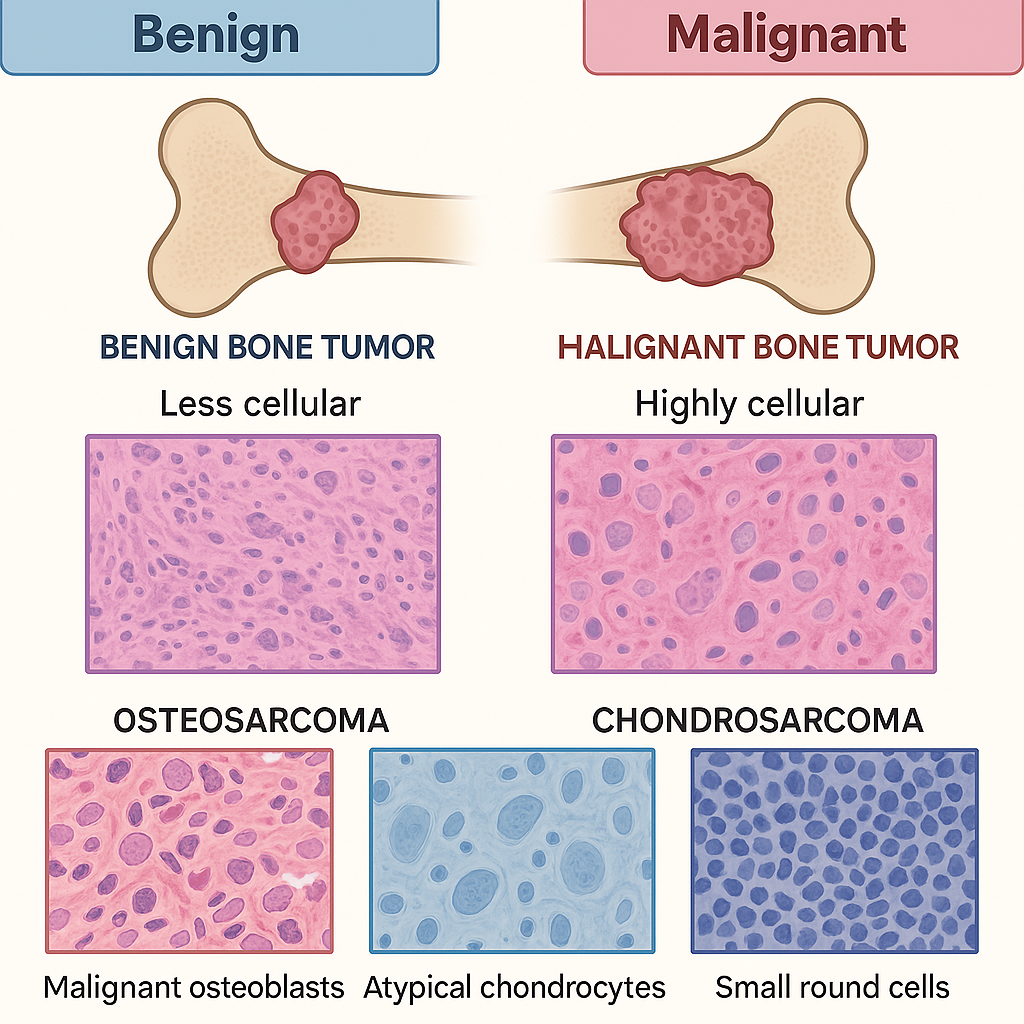
🤲 Rheumatoid Arthritis – Pathological Changes
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease that primarily affects synovial joints, leading to inflammation, joint destruction, and functional disability. Unlike osteoarthritis (mechanical), RA is an inflammatory condition with autoimmune features.
🔬 I. Pathogenesis Overview
RA is triggered by an autoimmune response involving:
- Genetic susceptibility (e.g., HLA-DR4/DR1)
- Environmental triggers (e.g., infections, smoking)
- Autoantibodies:
- Rheumatoid Factor (RF)
- Anti-Citrullinated Protein Antibodies (ACPA)
These antibodies target synovial antigens, leading to chronic inflammation and structural changes.
🔥 II. Pathological Changes in RA (Joint Level)
🟡 1. Synovial Membrane Inflammation (Synovitis)
- In early RA, synovial cells become hyperplastic, expanding from 1–2 cell layers to 8–10 layers.
- Infiltration by:
- T cells (CD4+ helper), B cells, macrophages
- Plasma cells producing autoantibodies
- Leads to edema, warmth, and joint swelling.
🟠 2. Formation of Pannus
- The inflamed synovium grows into the joint cavity, forming a pannus:
- A thickened, vascular, invasive granulation tissue.
- Contains inflammatory cells, fibroblasts, and blood vessels.
- Pannus invades cartilage and bone, destroying joint structures.
🔴 3. Articular Cartilage Destruction
- The pannus releases enzymes:
- Matrix metalloproteinases (MMPs)
- Collagenases
- These degrade articular cartilage, eroding the smooth joint surface.
- Leads to joint space narrowing and stiffness.
⚫ 4. Bone Erosion
- Activated osteoclasts (stimulated by RANKL) resorb subchondral bone.
- Creates marginal erosions especially near joint margins.
- Bone loss contributes to joint instability and deformities.
🧬 III. Chronic Stage – Fibrosis and Ankylosis
🧱 5. Fibrosis
- Chronic inflammation leads to fibrosis of the synovium.
- Replacement of granulation tissue by fibrous connective tissue.
🦴 6. Fibrous or Bony Ankylosis
- In late disease:
- Fibrous ankylosis = fibrous tissue fuses joint
- Bony ankylosis = bone grows across joint space
- Results in permanent joint immobility.
🧠 Histological Features
| Feature | Description |
|---|---|
| Synovial hyperplasia | Thickened synovial lining with 8–10 layers |
| Lymphoid follicles | Resemble secondary lymphoid tissue |
| Pannus | Invasive fibrovascular tissue |
| Cartilage erosion | Loss of proteoglycans and collagen |
| Bone erosion | Osteoclast-mediated lytic lesions |
🩺 Extra-articular Pathological Changes
- Rheumatoid nodules: Granuloma-like lesions (central necrosis, palisading histiocytes)
- Vasculitis: Necrotizing small vessel inflammation
- Lung involvement: Interstitial fibrosis, pleuritis
- Heart: Pericarditis, myocarditis
- Eyes: Scleritis, keratoconjunctivitis sicca
🔄 Summary of Pathological Progression in RA
- Autoimmune synovitis → Synovial cell proliferation
- Pannus formation → cartilage invasion
- Cartilage and bone destruction
- Fibrosis and ankylosis
- Chronic deformity and loss of function
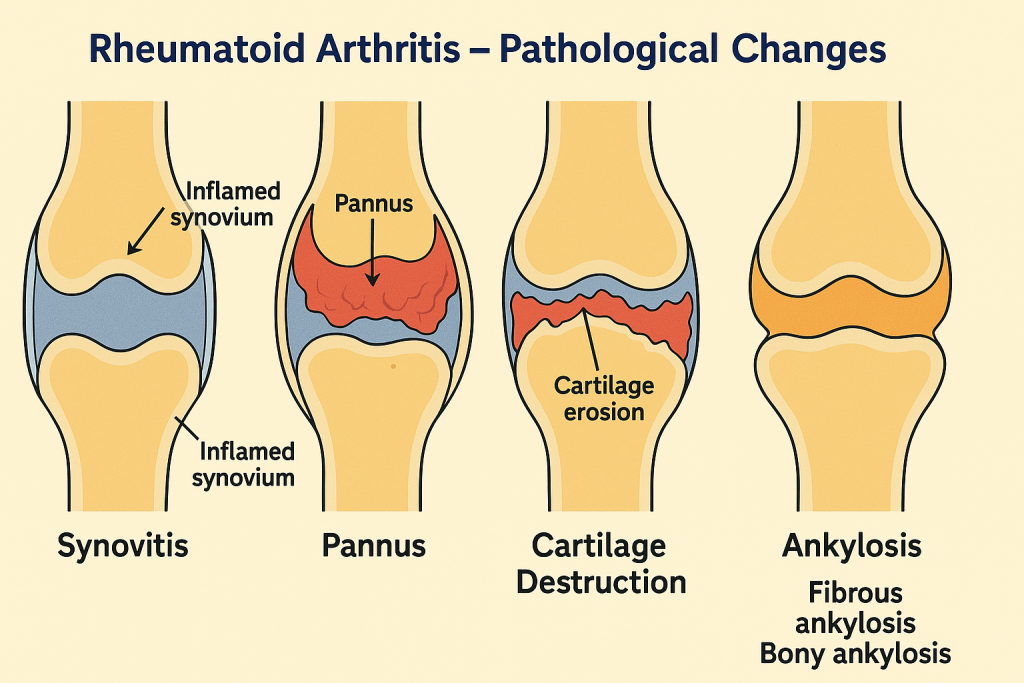
🦴 Osteoarthritis – Pathological Changes
Osteoarthritis (OA) is a chronic, degenerative joint disease primarily affecting articular cartilage, characterized by the progressive breakdown of joint structures and non-inflammatory synovial changes. It is the most common type of arthritis, especially in older adults, and is often considered a “wear and tear” arthritis.
Though previously thought to be purely mechanical, OA also involves low-grade inflammation, metabolic imbalance, and structural remodeling of joint components.
🔍 Pathogenesis Overview
Osteoarthritis begins with the loss of balance between cartilage degradation and synthesis. Risk factors like age, obesity, trauma, joint misalignment, and genetics accelerate this imbalance.
🔬 I. Pathological Changes in Articular Cartilage
🔸 1. Cartilage Matrix Degeneration
- Proteoglycans and collagen (especially type II) are lost.
- Chondrocytes respond by attempting repair but become dysfunctional.
- Cartilage becomes soft, fibrillated, and loses elasticity.
🔸 2. Surface Fibrillation and Cracks
- Cartilage surface develops fissures and vertical clefts.
- Fibrillation starts superficially and progresses into deeper layers.
🔸 3. Cartilage Thinning and Erosion
- Continued matrix degradation leads to progressive loss of cartilage thickness.
- Eventually, the cartilage is completely lost, leading to bone-on-bone contact.
🦴 II. Subchondral Bone Changes
🔸 4. Subchondral Sclerosis
- Bone beneath the cartilage (subchondral bone) becomes dense and sclerotic.
- Reflects attempted remodeling due to increased mechanical load.
🔸 5. Cyst Formation
- Microfractures in subchondral bone allow synovial fluid intrusion → subchondral cysts.
🔸 6. Osteophyte Formation
- New bone outgrowths (osteophytes) develop at joint margins.
- Represent a reactive attempt to stabilize the joint.
🧫 III. Synovial and Capsular Changes
🔸 7. Mild Synovitis
- Unlike RA, inflammation in OA is mild and secondary.
- Synovium shows mononuclear infiltration, hyperplasia, and fibrosis.
🔸 8. Joint Capsule Thickening
- Fibrosis and remodeling lead to capsular thickening and joint stiffness.
🦵 IV. Muscles and Ligament Involvement
- Ligaments become lax, contributing to joint instability.
- Muscle wasting occurs around affected joints due to disuse and pain.
🧠 Histological Features of OA
| Structure | Pathological Changes |
|---|---|
| Articular cartilage | Fibrillation, erosion, chondrocyte clusters |
| Subchondral bone | Sclerosis, cysts, remodeling |
| Joint margin | Osteophyte formation |
| Synovium | Mild chronic inflammation, fibrosis |
| Capsule/ligaments | Thickening, degeneration |
🔄 Summary of Pathological Sequence in OA
- Mechanical/cartilage stress
- Chondrocyte damage and matrix loss
- Cartilage fibrillation → erosion
- Subchondral bone sclerosis + cysts
- Osteophyte formation at margins
- Joint space narrowing, stiffness, and pain
🩺 Key Clinical Correlation
- Pain worsens with activity, relieved by rest.
- Crepitus, stiffness, reduced range of motion.
- Radiologic signs: Joint space narrowing, osteophytes, subchondral sclerosis, cysts
Endocrine system
🩸 Diabetes Mellitus – Pathological Changes
Diabetes Mellitus is a chronic metabolic disorder characterized by hyperglycemia, resulting from defects in insulin secretion, insulin action, or both. Over time, sustained high blood sugar causes widespread damage to various organ systems.
There are two main pathological types:
- 🔵 Type 1 DM: Autoimmune destruction of β-cells (absolute insulin deficiency)
- 🟠 Type 2 DM: Insulin resistance with relative insulin deficiency
Despite different origins, both types eventually lead to chronic hyperglycemia, which underlies most of the pathological changes.
🔬 I. Pancreatic Changes
🔵 Type 1 Diabetes Mellitus (T1DM)
- Insulitis: Lymphocytic infiltration of pancreatic islets.
- Destruction of β-cells in the islets of Langerhans.
- Islets become shrunken, fibrotic, and eventually disappear.
- Autoantibodies against insulin, GAD-65, and islet cell antigens.
🟠 Type 2 Diabetes Mellitus (T2DM)
- Islet hypertrophy in early stages.
- Amyloid deposition in and around islets (amylin accumulation).
- Mild to moderate β-cell loss.
- No insulitis; instead, mild inflammatory infiltration may be present.
🧬 II. Vascular Changes – Central in Chronic Complications
Chronic hyperglycemia induces glycation of proteins, oxidative stress, and inflammatory cascades, which damage endothelial cells.
🔹 A. Microvascular Changes (Small Vessels)
- Thickening of basement membranes (capillaries of retina, glomeruli, vasa nervorum)
- Hyaline arteriolosclerosis
- Leads to:
- Diabetic retinopathy
- Diabetic nephropathy
- Diabetic neuropathy
🔸 B. Macrovascular Changes (Large Vessels)
- Accelerated atherosclerosis due to:
- Endothelial dysfunction
- Dyslipidemia
- Pro-inflammatory state
- Involves:
- Coronary arteries → MI
- Cerebral arteries → Stroke
- Peripheral arteries → Gangrene, ulcers
🧠 III. Organ-Specific Pathological Changes
👁️ 1. Eye (Diabetic Retinopathy)
- Non-proliferative stage:
- Microaneurysms, retinal hemorrhages, cotton wool spots
- Proliferative stage:
- Neovascularization → Risk of retinal detachment and blindness
🧠 2. Nervous System (Diabetic Neuropathy)
- Axonal degeneration and demyelination
- Affects sensory, motor, and autonomic nerves
- Vasa nervorum show ischemic changes
🩺 3. Kidney (Diabetic Nephropathy)
- Glomerular basement membrane thickening
- Mesangial expansion
- Kimmelstiel-Wilson nodules (nodular glomerulosclerosis)
- Proteinuria → nephrotic syndrome → ESRD
💉 4. Skin and Immune System
- Poor wound healing
- Increased susceptibility to infections (fungal, bacterial)
- Necrobiosis lipoidica (in rare cases)
🔄 Summary Table – Pathological Features in Diabetes Mellitus
| System | Key Pathological Changes |
|---|---|
| Pancreas | β-cell destruction (T1), islet amyloid (T2) |
| Vessels | Basement membrane thickening, atherosclerosis |
| Eye | Retinal microaneurysms, neovascularization |
| Kidney | Glomerulosclerosis, proteinuria, Kimmelstiel-Wilson lesions |
| Nerves | Axonal degeneration, ischemia of vasa nervorum |
| Heart | Coronary artery disease |
| Skin/Immune | Impaired immunity, poor healing, infection susceptibility |
🌟 Pathogenesis Highlights of Chronic Hyperglycemia
- Advanced Glycation End Products (AGEs): Cross-linking of proteins damaging vessels
- Polyol pathway: Sorbitol accumulation in nerves and lens → damage
- Protein kinase C activation: Alters blood flow and increases inflammation
🦋 Goitre – Pathological Changes
Goitre refers to an abnormal enlargement of the thyroid gland, regardless of thyroid hormone function. It may occur in euthyroid, hypothyroid, or hyperthyroid states.
Goitres are broadly classified into:
- 🔹 Diffuse Goitre
- 🔸 Nodular Goitre (Multinodular or Solitary)
- 🟠 Toxic vs. Non-toxic
The underlying pathological changes depend on the cause — commonly iodine deficiency, autoimmune disorders, or hormonal dysregulation.
🔬 I. Pathogenesis Overview
The development of a goitre typically involves:
- Increased TSH stimulation (due to low thyroid hormones or iodine deficiency)
- Hyperplasia and hypertrophy of thyroid follicular cells
- Over time: nodularity, fibrosis, hemorrhage, and cystic degeneration
🧬 II. Pathological Changes by Stages
1. Diffuse Hyperplastic (Physiological or Early Simple Goitre)
- Seen in early iodine deficiency or puberty/pregnancy.
- Uniform enlargement of the gland.
- Microscopically:
- Follicular epithelial hypertrophy (columnar cells)
- Scalloping of colloid due to increased reabsorption
- Follicles may be small with reduced colloid
2. Colloid Goitre (Endemic/Simple)
- Occurs in iodine-deficient areas.
- Follicles distended with abundant colloid
- Lining epithelium becomes flattened due to inactivity.
- Grossly: Gland is smooth, soft, and rubbery
3. Multinodular Goitre (Long-standing Simple Goitre)
- Results from repeated cycles of hyperplasia and involution
- Gland becomes:
- Asymmetrically enlarged
- Lobulated and nodular
- Histological features:
- Irregular follicles of varying sizes
- Areas of hemorrhage, fibrosis, calcification, and cystic change
- Nodules may show colloid-rich or hyperplastic follicles
- Some regions may resemble adenomatous hyperplasia
⚠️ Potential Complications of Multinodular Goitre
- Compression symptoms: dysphagia, dyspnea, hoarseness
- Toxic nodular goitre: Some nodules may become autonomously functioning → hyperthyroidism
- Malignant transformation (rare): especially in long-standing goitres
🧠 Special Types of Goitre – Pathological Variants
🟢 Hashimoto’s Thyroiditis (Autoimmune Goitre)
- Diffuse enlargement, initially firm, later atrophic
- Histology:
- Lymphocytic infiltration with germinal centers
- Hurthle cell metaplasia
- Fibrosis in late stage
🔴 Graves’ Disease (Toxic Diffuse Goitre)
- Gland is diffusely enlarged and vascular
- Follicles show:
- Tall columnar epithelium
- Scalloped colloid
- Lymphoid infiltration with activated follicles
🔄 Summary of Pathological Progression
| Stage/Type | Pathological Changes |
|---|---|
| Diffuse hyperplastic | Follicular hypertrophy, reduced colloid |
| Colloid goitre | Follicles distended with colloid, flattened epithelium |
| Multinodular goitre | Irregular follicles, fibrosis, hemorrhage, calcification |
| Toxic goitre | Functional autonomy, possible hyperthyroidism |
| Autoimmune goitre | Lymphocytic infiltration, Hurthle cell change |
🦋 Thyroid Carcinoma – Pathological Changes
Thyroid carcinoma refers to malignant tumors arising from the epithelial cells of the thyroid gland. It is the most common endocrine malignancy and includes several distinct histopathological types, each with unique morphological and clinical characteristics.
The major types of thyroid carcinoma include:
- 🔹 Papillary Carcinoma (PTC) – Most common
- 🔸 Follicular Carcinoma (FTC)
- 🟣 Medullary Carcinoma (MTC)
- 🔴 Anaplastic Carcinoma (ATC) – Most aggressive
🔬 I. Papillary Thyroid Carcinoma (PTC)
🌟 Most common type (80–85%) – usually well-differentiated
🔸 Gross Features:
- Usually single or multifocal, firm, whitish tumor
- May show calcification or cystic changes
🔬 Microscopic Pathology:
- Papillary structures with fibrovascular cores
- Nuclear features are hallmark:
- “Orphan Annie eye” nuclei (empty-appearing chromatin)
- Nuclear grooves
- Intranuclear pseudoinclusions
- May contain psammoma bodies (calcified concentric layers)
- Lymphatic invasion is common → cervical lymph node metastasis
🧬 II. Follicular Thyroid Carcinoma (FTC)
🌾 Second most common – encapsulated, often misdiagnosed as adenoma
🔸 Gross Features:
- Well-circumscribed, tan-brown mass
- Often encapsulated
🔬 Microscopic Pathology:
- Uniform small follicles resembling normal thyroid
- Diagnostic hallmark: capsular and/or vascular invasion
- No papillary nuclear features
- Hematogenous spread (to lungs, bones) is more common than lymphatic
🧪 III. Medullary Thyroid Carcinoma (MTC)
🧬 Arises from parafollicular C cells, not follicular cells
🔸 Gross Features:
- Solid, firm, gray or pale tumors
- May show hemorrhage or necrosis
🔬 Microscopic Pathology:
- Sheets or nests of polygonal to spindle-shaped cells
- Amyloid stroma (from calcitonin deposition) – Congo red positive
- Cells produce calcitonin (used for diagnosis)
- May be associated with MEN 2A/2B syndromes
🔥 IV. Anaplastic Thyroid Carcinoma (ATC)
💀 Highly aggressive, undifferentiated carcinoma
🔸 Gross Features:
- Large, bulky, invasive tumor
- Frequently invades trachea, esophagus, and major neck vessels
🔬 Microscopic Pathology:
- Highly pleomorphic giant and spindle cells
- Frequent mitoses, necrosis, and vascular invasion
- No features of normal thyroid architecture
- Often arises from dedifferentiation of PTC or FTC
🧠 Summary Table – Pathological Features of Thyroid Carcinomas
| Type | Cell Origin | Key Features | Spread |
|---|---|---|---|
| Papillary (PTC) | Follicular cells | Papillae, nuclear clearing, grooves, psammoma | Lymphatic |
| Follicular (FTC) | Follicular cells | Follicles, capsular/vascular invasion | Hematogenous |
| Medullary (MTC) | Parafollicular C | Calcitonin, amyloid stroma, neuroendocrine look | Lymphatic & blood |
| Anaplastic (ATC) | Dedifferentiated | Pleomorphic cells, aggressive, necrotic | Direct invasion |
🚨 Common Pathological Outcomes
- Capsular and vascular invasion → metastasis
- Local invasion → airway obstruction, dysphagia, hoarseness
- Hormonal effects:
- PTC/FTC: usually euthyroid
- MTC: may have diarrhea, flushing due to calcitonin and peptides