BSC SEM 3 UNIT 8 ADULT HEALTH NURSING 1
UNIT 8 Nursing management of patients with disorders of endocrine system
🧠 Review of Anatomy and Physiology of the Endocrine System
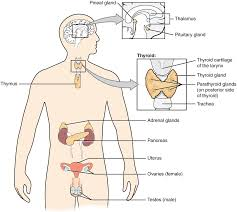
📌 Definition of Endocrine System:
The endocrine system is a network of glands that produce and secrete hormones directly into the bloodstream to regulate the body’s growth, metabolism, development, tissue function, reproduction, mood, and homeostasis.
⚙️ Major Characteristics of the Endocrine System:
| 🧪 Feature | 💡 Description |
|---|---|
| 🔄 Regulation | Maintains long-term processes like growth and development |
| 🧬 Hormone-based | Uses chemical messengers called hormones |
| 🧠 Close integration with nervous system | Works with hypothalamus to coordinate body functions |
| ⏳ Slower but prolonged response | Compared to nervous system (which is faster but short-lived) |
🧍♂️ Major Endocrine Glands and Hormones:
| 🔬 Gland | 📍 Location | 🌟 Hormones Secreted | 📋 Function |
|---|---|---|---|
| Hypothalamus | Brain (below thalamus) | CRH, TRH, GnRH, GHRH, Somatostatin | Regulates pituitary gland |
| Pituitary (Master Gland) | Base of brain | Anterior: GH, TSH, ACTH, FSH, LH, PRL Posterior: ADH, Oxytocin | Regulates other endocrine glands |
| Pineal gland | Brain (epithalamus) | Melatonin | Controls circadian rhythm (sleep-wake cycle) |
| Thyroid gland | Neck (anterior to trachea) | T3, T4, Calcitonin | Regulates metabolism, growth, and calcium balance |
| Parathyroid glands | Behind thyroid (4 small glands) | Parathyroid hormone (PTH) | Raises blood calcium levels |
| Adrenal glands | On top of kidneys | Cortex: Cortisol, Aldosterone Medulla: Adrenaline, Noradrenaline | Stress response, BP, metabolism |
| Pancreas (dual role) | Abdomen (behind stomach) | Insulin, Glucagon, Somatostatin | Regulates blood sugar levels |
| Gonads (Ovaries/Testes) | Pelvic region | Estrogen, Progesterone, Testosterone | Reproduction and secondary sex characteristics |
| Thymus (in children) | Upper chest (behind sternum) | Thymosin | Promotes T-cell development (immune role) |
🧪 Hormones – Nature and Types:
| 🧬 Type | 🧾 Examples | 💡 Characteristics |
|---|---|---|
| Peptide hormones | Insulin, ADH, GH | Water-soluble, act via receptors on cell surface |
| Steroid hormones | Cortisol, Estrogen, Testosterone | Lipid-soluble, act on intracellular receptors |
| Amino acid derivatives | T3, T4, Adrenaline | Can be water- or lipid-soluble |
🔁 Mechanism of Hormone Action:
- Endocrine gland secretes hormone into bloodstream
- Hormone travels to target organ/cells
- Binds to specific receptor (cell membrane or nucleus)
- Triggers biological response (gene expression, enzyme activation, etc.)
- Negative feedback loop controls further secretion (e.g., TSH → T3/T4 → inhibits TSH)
🔄 Feedback Control:
| 🔄 Type | 🔁 Description | 📌 Example |
|---|---|---|
| ❌ Negative Feedback | Stops hormone production when desired effect is reached | TSH–T3/T4 axis |
| ➕ Positive Feedback | Enhances hormone secretion until an event completes | Oxytocin in labor |
⚠️ Endocrine vs. Exocrine Glands:
| Feature | Endocrine | Exocrine |
|---|---|---|
| Ducts | Ductless | Has ducts |
| Secretion | Into blood | Into body surface/cavity |
| Examples | Pituitary, Thyroid | Salivary, Sweat glands |
🩺 Physiological Roles of Hormones:
| 🔍 Function | 🧬 Hormones Involved |
|---|---|
| 🧠 Growth & Development | GH, Thyroid hormones |
| 🔥 Metabolism | T3, T4, Insulin, Glucagon |
| 🧃 Fluid & Electrolyte balance | ADH, Aldosterone |
| 🩸 Blood glucose control | Insulin (↓), Glucagon (↑) |
| 🫀 Stress response | Cortisol, Adrenaline |
| 🧬 Reproduction | Estrogen, Progesterone, Testosterone, LH, FSH |
💉 Clinical Relevance / Disorders:
| 🩺 Disorder | 🧬 Affected Gland | 🔄 Hormonal Imbalance |
|---|---|---|
| Diabetes Mellitus | Pancreas | ↓ Insulin |
| Hypothyroidism | Thyroid | ↓ T3, T4 |
| Cushing’s Syndrome | Adrenal Cortex | ↑ Cortisol |
| Acromegaly | Pituitary (anterior) | ↑ GH in adults |
| Diabetes Insipidus | Posterior Pituitary | ↓ ADH |
| Addison’s Disease | Adrenal Cortex | ↓ Cortisol, Aldosterone |
🧷 Key Points:
✅ Endocrine system controls long-term body functions through hormones
✅ Hormones act on specific target organs via receptors
✅ Maintains homeostasis, growth, metabolism, stress response, and reproduction
✅ Feedback mechanisms maintain hormonal balance
✅ Disorders often arise due to hormone deficiency or excess
🩺 Nursing Assessment of Patients with Disorders of the Endocrine System
📌 Purpose of Assessment:
To identify signs and symptoms of hormonal imbalances, determine functional changes, evaluate the impact on body systems, and guide effective nursing and medical interventions.
🔍 I. Health History Collection
1. 🧬 General Information:
- Age, sex, weight changes
- Chief complaints: fatigue, weight loss/gain, polyuria, polydipsia, etc.
2. 🩺 Presenting Symptoms:
Ask about:
- Fatigue, weakness
- Intolerance to heat or cold
- Hair loss or excessive hair growth
- Skin dryness or pigmentation
- Menstrual irregularities, infertility
- Changes in libido or sexual function
- Memory issues, depression, or anxiety
3. 🧬 Past Medical History:
- Previous endocrine disorders (e.g., diabetes, thyroid disease)
- Radiation or surgery to the head, neck, or abdomen
- Autoimmune disorders
- History of head trauma or brain surgery
4. 👨👩👧👦 Family History:
- Genetic endocrine conditions (e.g., diabetes, thyroid cancer)
5. 💊 Medication History:
- Hormonal therapy (thyroxine, insulin, steroids)
- Use of oral contraceptives or androgens
- Over-the-counter supplements or herbal remedies
6. 💼 Lifestyle and Social History:
- Dietary habits, physical activity
- Stress levels and coping mechanisms
- Alcohol, tobacco, or drug use
👁️ II. Physical Examination
A systematic head-to-toe examination is crucial.
1. 🧠 General Appearance:
- Body build, stature, obesity (central/peripheral)
- Facial expression (moon face, facial puffiness, exophthalmos)
2. 🌡️ Vital Signs:
- Temperature (fever in hyperthyroidism, cold intolerance in hypothyroidism)
- Heart rate and rhythm (tachycardia, bradycardia, arrhythmias)
- Respiratory rate and BP (hypertension in Cushing’s, low BP in Addison’s)
3. 🦴 Skin and Hair:
- Dryness, thinning, pigmentation, bruising
- Hair loss (alopecia) or hirsutism
- Acne or seborrhea
4. 👁️ Eyes:
- Exophthalmos (thyroid disorders)
- Visual disturbances
5. 💬 Neck (Thyroid Gland):
- Palpate for size, tenderness, nodules
- Observe for goiter or surgical scars
6. 🫀 Cardiovascular System:
- Pulse (bounding in hyperthyroidism)
- BP variations (orthostatic hypotension in Addison’s)
7. 🧃 Fluid Balance:
- Signs of dehydration or edema
- Weight gain/loss
8. 🦴 Musculoskeletal System:
- Muscle wasting or weakness
- Tetany or cramps (calcium imbalance)
9. 🧠 Neurological Assessment:
- Orientation, memory, mood
- Reflexes (hyperreflexia in hyperthyroidism, sluggish reflexes in hypothyroidism)
10. 🧬 Reproductive System:
- Menstrual pattern
- Erectile dysfunction, fertility issues
🧪 III. Diagnostic Investigations (Reviewed by Nurse):
| 🧾 Test | 🔍 Purpose |
|---|---|
| ✅ Blood glucose | Detect diabetes mellitus |
| ✅ Thyroid panel (TSH, T3, T4) | Assess thyroid function |
| ✅ Serum cortisol (AM/PM) | Evaluate adrenal function |
| ✅ ACTH stimulation test | For Addison’s or Cushing’s |
| ✅ Serum calcium/phosphate | Parathyroid function |
| ✅ HbA1c | Long-term glycemic control |
| ✅ Urinary catecholamines/metanephrines | For pheochromocytoma |
| ✅ MRI/CT | Pituitary, adrenal, or thyroid gland imaging |
🗂️ IV. Nursing Assessment Tools:
| 📋 Tool | 📌 Used For |
|---|---|
| ✅ Glucometer | Monitor blood glucose levels |
| ✅ Weight chart | Detect weight loss/gain trends |
| ✅ Intake-output chart | Monitor fluid/electrolyte balance |
| ✅ Pain scale | Assess discomfort due to neuropathy or gland enlargement |
| ✅ Neurological scale (GCS/MMSE) | Monitor altered sensorium |
🧷 Key Nursing Assessment Points by Common Disorders:
| 🔍 Disorder | 🩺 Focused Nursing Assessment |
|---|---|
| Diabetes Mellitus | Blood glucose, wound healing, vision, sensation, hydration |
| Hypothyroidism | Cold intolerance, dry skin, constipation, bradycardia |
| Hyperthyroidism | Heat intolerance, weight loss, tremors, anxiety, tachycardia |
| Cushing’s Syndrome | Moon face, buffalo hump, striae, hyperglycemia |
| Addison’s Disease | Fatigue, hypotension, hyperpigmentation, dehydration |
| Pheochromocytoma | Severe hypertension, palpitations, headache, sweating |
🧷 Key Points:
✅ Accurate history and physical exam are vital in identifying endocrine disorders
✅ Watch for subtle signs: mood changes, fatigue, weight change, hair/skin changes
✅ Always assess lab values and correlate clinically
✅ Monitor for potential complications like hypoglycemia, thyroid storm, adrenal crisis
✅ Holistic and patient-centered assessment ensures better outcomes
🩺 History and Physical Assessment of Patients with Endocrine Disorders
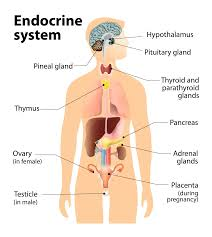
📌 Purpose:
To identify dysfunction in hormone production, regulation, or target tissue response and to detect systemic effects of endocrine imbalance. Assessment helps in early detection, accurate diagnosis, and effective management.
🗂️ I. Comprehensive Health History
A detailed history provides critical clues about the type and extent of hormonal dysfunction. Use open-ended and focused questions.
🧾 A. Chief Complaint (CC):
Ask the patient:
➡️ “What brings you here today?”
Typical complaints:
- Fatigue
- Weight loss/gain
- Changes in appetite or thirst
- Irregular menses or libido changes
- Palpitations
- Mood changes
- Increased urination
🧠 B. History of Present Illness (HPI):
Clarify:
- Onset: Sudden or gradual
- Duration: Days, weeks, months
- Severity: Intensity of symptoms
- Aggravating/Relieving Factors: Triggers (e.g., stress, food)
- Associated Symptoms: Cold/heat intolerance, visual changes, skin or hair changes
📅 C. Past Medical History (PMH):
Check for:
- Previous endocrine disorders (DM, thyroid dysfunction, pituitary tumor)
- Radiation therapy to head/neck
- Head injury or brain surgery
- Autoimmune diseases (e.g., SLE, rheumatoid arthritis)
👨👩👧👦 D. Family History:
Ask about hereditary endocrine conditions:
- Diabetes mellitus
- Thyroid cancer or goiter
- Multiple endocrine neoplasia (MEN) syndromes
💊 E. Medication History:
Include:
- Hormone replacement therapy (e.g., insulin, thyroxine, steroids)
- Long-term corticosteroid use
- Birth control pills
- Herbal supplements (may affect hormones)
🧬 F. Personal and Social History:
Assess:
- Stress levels
- Diet and lifestyle (high sugar/salt/fat intake)
- Smoking, alcohol, drug use
- Exercise habits
- Occupational exposure (e.g., radiation, chemicals)
🚺 G. Reproductive History (for women):
- Menstrual pattern
- Fertility history
- Menopause status
- Use of hormone therapies
🧍♀️ II. Physical Assessment: A Head-to-Toe Approach
Focus on general appearance, glandular swelling, skin/hair changes, and systemic symptoms.
🧠 A. General Observation:
- Facial appearance: Moon face, mask-like face, exophthalmos
- Body build: Central obesity (Cushing’s), lean build (hyperthyroidism)
- Gait and posture
- Mental status: Depression, anxiety, confusion
🌡️ B. Vital Signs:
| 🩺 Vital Sign | 💡 Relevance |
|---|---|
| BP | Hypotension (Addison’s), hypertension (Cushing’s, pheochromocytoma) |
| Pulse | Bradycardia (hypothyroidism), tachycardia (hyperthyroidism) |
| Temperature | Fever (thyroid storm), low temp (hypothyroid) |
| Weight | Sudden weight loss/gain |
👀 C. Head and Neck:
- Eyes: Exophthalmos, blurred vision (Graves’ disease)
- Thyroid gland: Palpate for size, nodules, tenderness, thrill/bruit
- Skin: Dryness (hypothyroidism), thinning, acne, pigmentation, bruising
🦴 D. Musculoskeletal System:
- Muscle wasting or weakness
- Joint pain
- Short stature or delayed growth in children
- Bone tenderness (hyperparathyroidism)
🧃 E. Fluid and Electrolyte Balance:
- Dehydration signs (dry mucosa, poor skin turgor)
- Edema (myxedema in hypothyroidism)
- Polyuria, polydipsia (diabetes)
🧠 F. Neurological Status:
- Reflexes (hyperreflexia in hyperthyroidism, slow in hypothyroidism)
- Orientation, memory, behavior
- Neuropathy (numbness, tingling in diabetes)
- Seizures (hypocalcemia)
❤️ G. Cardiovascular and Respiratory:
- Palpitations, irregular heart rhythms
- Chest pain or breathlessness
- Heart sounds (pericardial effusion in hypothyroidism)
🧬 H. Abdominal Examination:
- Distension (ascites in Cushing’s)
- Liver enlargement (fatty liver in diabetes)
- Adrenal mass (pheochromocytoma)
🚺 I. Reproductive and Genitourinary:
- Amenorrhea or oligomenorrhea
- Erectile dysfunction
- Infertility
- Changes in libido
📊 III. Functional and Diagnostic Assessment (Reviewed by Nurse):
| 🔍 Test | Purpose |
|---|---|
| ✅ Blood glucose (FBS/RBS/HbA1c) | Diabetes diagnosis/control |
| ✅ Thyroid profile (TSH, T3, T4) | Thyroid dysfunction |
| ✅ Cortisol (AM/PM) | Adrenal function |
| ✅ ACTH stimulation test | Adrenal insufficiency |
| ✅ Serum electrolytes | Na⁺, K⁺, Ca²⁺ imbalance |
| ✅ MRI/CT | Tumors (pituitary, adrenal) |
| ✅ Urine tests | 24-hr catecholamines, ketones |
🧷 Key Points:
✅ Always correlate subjective complaints with objective findings
✅ Endocrine disorders can have multi-systemic effects – assess holistically
✅ Monitor trends in weight, energy levels, mental state, and skin/hair changes
✅ Early detection and documentation help prevent complications like thyroid storm, myxedema coma, or adrenal crisis
✅ Nurses play a critical role in ongoing monitoring, patient education, and early warning sign identification
🦋 Disorders of the Thyroid Gland
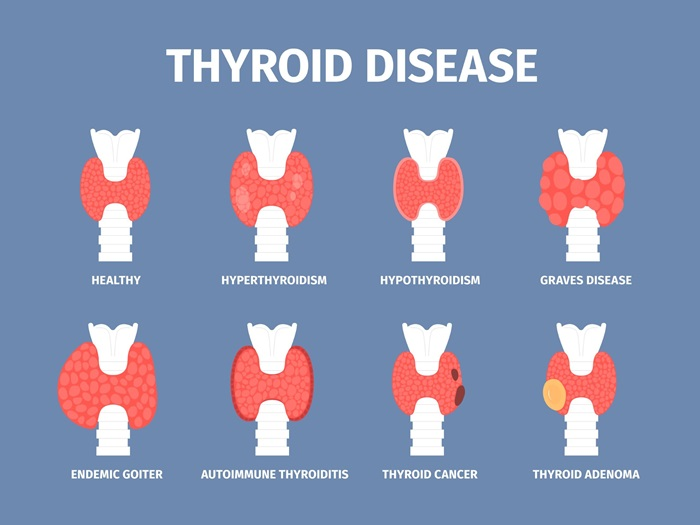
📌 Overview of the Thyroid Gland:
- The thyroid is a butterfly-shaped gland located anterior to the trachea in the neck.
- It secretes:
- T3 (Triiodothyronine)
- T4 (Thyroxine)
- Calcitonin (involved in calcium regulation)
- Controlled by the Hypothalamic–Pituitary–Thyroid Axis:
- Hypothalamus → TRH
- Pituitary → TSH
- Thyroid → T3 & T4
⚠️ Common Disorders of the Thyroid Gland:
| 🔍 Disorder | ⬆️/⬇️ Function | 💡 Description |
|---|---|---|
| Hypothyroidism | ↓ | Underactive thyroid hormone production |
| Hyperthyroidism | ↑ | Overactive thyroid hormone production |
| Goiter | Variable | Enlargement of the thyroid gland |
| Thyroiditis | ↓ or ↑ | Inflammation of the thyroid gland |
| Thyroid nodules/cysts | Variable | Lumps in the thyroid; benign or malignant |
| Thyroid cancer | Variable | Malignancy of thyroid tissue |
🧪 1. Hypothyroidism
✅ Definition:
A condition in which the thyroid gland fails to produce sufficient T3 and T4, slowing down body metabolism.
🎯 Causes:
- Autoimmune (e.g., Hashimoto’s thyroiditis)
- Iodine deficiency
- Post-thyroidectomy or radioactive iodine treatment
- Congenital (cretinism)
- Drug-induced (e.g., lithium, amiodarone)
🧠 Signs & Symptoms:
- Fatigue, weight gain, cold intolerance
- Constipation, depression, dry skin, brittle hair
- Bradycardia, slow reflexes
- Menstrual irregularities
- Myxedema (severe form)
🔬 Diagnosis:
- ↑ TSH, ↓ T3 & T4 (Primary hypothyroidism)
- ↓ TSH, ↓ T3 & T4 (Secondary hypothyroidism)
- Thyroid antibodies in autoimmune cases
💊 Management:
- Hormone replacement: Levothyroxine
- Lifelong therapy with regular monitoring
🔥 2. Hyperthyroidism
✅ Definition:
Excess production of thyroid hormones, accelerating metabolic rate.
🎯 Causes:
- Graves’ disease (autoimmune, most common)
- Toxic multinodular goiter
- Thyroid adenoma
- Thyroiditis
🧠 Signs & Symptoms:
- Weight loss, increased appetite, heat intolerance
- Palpitations, tachycardia, nervousness
- Tremors, irritability, insomnia
- Exophthalmos (in Graves’ disease)
- Goiter
🔬 Diagnosis:
- ↓ TSH, ↑ T3 & T4
- Radioactive iodine uptake test
- TSH receptor antibodies (TRAb)
💊 Management:
- Antithyroid drugs: Methimazole, Propylthiouracil (PTU)
- Beta-blockers: Symptom control (e.g., propranolol)
- Radioactive iodine therapy
- Surgery: Subtotal or total thyroidectomy
🧊 3. Goiter
✅ Definition:
Enlargement of the thyroid gland, may be diffuse or nodular, associated with hypo-, hyper-, or euthyroid state.
🎯 Causes:
- Iodine deficiency (endemic goiter)
- Graves’ disease or Hashimoto’s thyroiditis
- Thyroid nodules
🧠 Symptoms:
- Neck swelling
- Difficulty swallowing/breathing (large goiter)
- Hoarseness
🔬 Diagnosis:
- Thyroid function tests (TFTs)
- Ultrasound or thyroid scan
💊 Management:
- Based on function (hyper/hypo)
- Iodine supplementation (if deficient)
- Surgery if compressive symptoms present
🔥 4. Thyroiditis
✅ Definition:
Inflammation of the thyroid, may be acute, subacute, or chronic.
🎯 Types:
- Hashimoto’s thyroiditis: Autoimmune, chronic, common in hypothyroidism
- Subacute (De Quervain’s): Viral, painful
- Postpartum thyroiditis
- Suppurative (bacterial): Rare
🧠 Symptoms:
- Neck pain (in subacute)
- Fever, malaise
- Transient hyperthyroidism → hypothyroidism
🔬 Diagnosis:
- ESR, CRP elevated (inflammation)
- TSH, T3, T4 levels
- Thyroid antibodies
💊 Management:
- NSAIDs, corticosteroids for inflammation
- Levothyroxine (if hypothyroid)
🦠 5. Thyroid Nodules and Cysts
✅ Definition:
Lumps in the thyroid gland, can be solid or fluid-filled (cystic).
🎯 Causes:
- Benign adenomas
- Colloid nodules
- Thyroid cancer
🧠 Symptoms:
- Usually asymptomatic
- Large nodules may cause neck pressure or hoarseness
🔬 Diagnosis:
- Ultrasound
- Fine needle aspiration (FNA) biopsy
- Thyroid scan (cold or hot nodules)
💊 Management:
- Observation if benign
- Surgery if malignant or compressive
🧬 6. Thyroid Cancer
✅ Types:
- Papillary carcinoma – Most common, slow-growing
- Follicular carcinoma – Moderate prognosis
- Medullary carcinoma – Genetic, associated with MEN syndrome
- Anaplastic carcinoma – Rare, aggressive
🧠 Symptoms:
- Painless neck mass
- Hoarseness, difficulty swallowing
- Enlarged cervical lymph nodes
🔬 Diagnosis:
- FNA biopsy
- Ultrasound, CT/MRI
- Serum calcitonin (medullary)
💊 Management:
- Surgery: Total thyroidectomy
- Radioactive iodine therapy
- Thyroxine suppression therapy
- Radiation/chemotherapy (for aggressive forms)
🩺 Nursing Considerations for Thyroid Disorders:
🔹 Assessment:
- Monitor vital signs (esp. heart rate, BP, temperature)
- Observe for signs of hypo- or hyperthyroidism
- Assess weight, energy, bowel habits, skin condition
🔹 Post-thyroidectomy Care:
- Monitor airway for obstruction or stridor
- Check for bleeding at surgical site
- Watch for hypocalcemia (Trousseau’s & Chvostek’s signs)
- Voice changes (recurrent laryngeal nerve damage)
🔹 Patient Education:
- Lifelong medication adherence
- Signs of under/overdose
- Importance of follow-up and monitoring
- Diet: Avoid goitrogens (e.g., cabbage, soy) in iodine-deficient patients
⚠️ Complications of Untreated Thyroid Disorders:
| 🛑 Disorder | 🚨 Complication |
|---|---|
| Hypothyroidism | Myxedema coma (life-threatening) |
| Hyperthyroidism | Thyroid storm (acute crisis) |
| Goiter | Tracheal compression |
| Thyroid cancer | Metastasis, airway obstruction |
| Thyroid surgery | Hypocalcemia, voice changes |
🧷 Key Points:
✅ Thyroid gland regulates metabolism, growth, and calcium balance
✅ Disorders include hypo-, hyperthyroidism, goiter, nodules, thyroiditis, and cancer
✅ Diagnosis is based on hormone levels, imaging, and biopsy
✅ Treatment includes medications, radioactive iodine, or surgery
✅ Nursing care focuses on assessment, monitoring complications, post-op care, and education
🦋 Hypothyroidism

📌 Definition:
Hypothyroidism is a clinical condition that results from the underproduction of thyroid hormones (T3 and T4) by the thyroid gland, leading to a slowing of metabolic processes in the body. It may be mild (subclinical) or severe (myxedema).
⚠️ Causes of Hypothyroidism:
🔹 A. Primary Hypothyroidism
(Problem is in the thyroid gland itself)
Most common form.
| 🚨 Cause | 📋 Details |
|---|---|
| Autoimmune thyroiditis | Hashimoto’s thyroiditis – most common in developed countries |
| Iodine deficiency | Most common cause globally (especially in endemic regions) |
| Thyroid surgery | Partial or total thyroidectomy |
| Radioactive iodine therapy | Used for hyperthyroidism, can cause thyroid damage |
| Congenital hypothyroidism | Born without a fully functioning thyroid |
| Drugs | Lithium, amiodarone, interferon-alpha |
| Infiltrative diseases | Sarcoidosis, hemochromatosis affecting thyroid |
🔹 B. Secondary Hypothyroidism
(Problem in the pituitary gland)
| 🚨 Cause | 📋 Details |
|---|---|
| Pituitary tumors | Compress or destroy TSH-secreting cells |
| Pituitary surgery/radiation | Causes hormonal imbalance |
| Sheehan’s syndrome | Postpartum pituitary infarction |
🔹 C. Tertiary Hypothyroidism
(Problem in the hypothalamus)
| 🚨 Cause | 📋 Details |
|---|---|
| Hypothalamic tumors | Interfere with TRH production |
| Trauma/inflammation | CNS infections, radiation injury |
🔹 D. Other Causes:
- Severe illness (euthyroid sick syndrome)
- Resistance to thyroid hormone (rare genetic condition)
🧬 Types of Hypothyroidism:
| 🏷️ Type | 📖 Description |
|---|---|
| Primary Hypothyroidism | Most common; due to direct failure of the thyroid gland |
| Secondary Hypothyroidism | Due to insufficient TSH secretion from the pituitary |
| Tertiary Hypothyroidism | Due to lack of TRH secretion from the hypothalamus |
| Congenital Hypothyroidism | Present at birth; can cause cretinism if untreated |
| Subclinical Hypothyroidism | Mild; ↑ TSH but normal T3 & T4 levels; asymptomatic or subtle signs |
| Overt Hypothyroidism | Full-blown symptoms with ↑ TSH and ↓ T3/T4 levels |
| Myxedema | Severe, life-threatening hypothyroidism with altered mental status, hypothermia, and multi-organ failure |
🔁 Pathophysiology of Hypothyroidism:
- 🧠 Disruption in the Hypothalamic–Pituitary–Thyroid (HPT) Axis:
- Normally:
- Hypothalamus secretes TRH → Stimulates pituitary to release TSH → TSH stimulates thyroid to produce T3 & T4.
- In hypothyroidism:
- Due to gland failure, TSH may rise (in primary) but T3/T4 remains low.
- Normally:
- 🔻 Reduced Thyroid Hormone Production:
- Decreased T3 (active) and T4 levels → Slow down cellular metabolism.
- 🐢 Slowing of Metabolic Processes:
- Reduced oxygen consumption and heat production
- Decreased energy utilization, protein synthesis, lipid metabolism
- 🔁 Feedback Mechanism Disrupted:
- Low T3/T4 → Pituitary increases TSH in primary hypothyroidism
- In secondary/tertiary, both TSH and T3/T4 are low
- ⚠️ Systemic Effects:
- Cardiovascular: Bradycardia, low cardiac output
- Nervous system: Slowed cognition, depression
- GI: Slowed motility → constipation
- Renal: Reduced GFR → fluid retention
- Skin: Dryness, thickening
- Hematologic: Anemia due to reduced erythropoietin
🚨 Signs and Symptoms of Hypothyroidism:
| 🔍 System | 🧾 Clinical Features |
|---|---|
| 🌡️ General | Fatigue, cold intolerance, weight gain despite poor appetite |
| 💆♀️ Skin/Hair | Dry, coarse skin; brittle nails; hair thinning or loss; puffy face |
| 💓 Cardiovascular | Bradycardia, hypotension, poor perfusion |
| 🧠 Neurological | Slow speech, depression, forgetfulness, drowsiness |
| 🍽️ Gastrointestinal | Constipation, anorexia, bloating |
| ♀️ Reproductive | Menorrhagia, infertility, low libido |
| 🦴 Musculoskeletal | Muscle weakness, cramps, joint stiffness |
| 👁️ Facial | Puffy eyes, hoarseness, macroglossia |
| 💨 Respiratory | Dyspnea on exertion, sleep apnea (in severe cases) |
| 🚨 Severe Case (Myxedema) | Hypothermia, coma, hypotension, respiratory depression |
🔔 Note: Symptoms are often gradual and nonspecific, especially in elderly patients.
🧪 Diagnosis of Hypothyroidism:
| 🧬 Test | 📌 Purpose / Interpretation |
|---|---|
| ✅ TSH (Thyroid-Stimulating Hormone) | Most sensitive test; ↑ in primary, ↓ in secondary/tertiary |
| ✅ Free T4 (Thyroxine) | ↓ in all forms of hypothyroidism |
| ✅ Free T3 (Triiodothyronine) | May be normal early on; ↓ in severe disease |
| ✅ Anti-TPO Antibodies | Positive in Hashimoto’s thyroiditis (autoimmune cause) |
| ✅ Thyroid Ultrasound | To assess size, nodules, inflammation |
| ✅ Radioactive Iodine Uptake (RAIU) | Low uptake in hypothyroidism |
| ✅ Lipid Profile | Often shows hypercholesterolemia and hypertriglyceridemia |
| ✅ CBC | May show normocytic or macrocytic anemia |
| ✅ ECG | May show sinus bradycardia, low voltage QRS |
💊 Medical Management of Hypothyroidism
🎯 Goal:
To restore and maintain normal thyroid hormone levels (euthyroid state) and manage associated metabolic derangements.
✅ 1. Hormone Replacement Therapy (Mainstay Treatment)
| 💊 Drug | 💡 Details |
|---|---|
| Levothyroxine (T4) | Drug of choice; synthetic form of thyroxine |
| Liothyronine (T3) | Occasionally used in myxedema coma or combination therapy |
📋 Levothyroxine – Administration Guidelines:
- 💊 Dose:
- Initial dose depends on age, weight, cardiac status, and severity of hypothyroidism
- Standard starting dose for adults: 50–100 mcg/day
- Lower dose (25–50 mcg/day) in elderly or cardiac patients
- 🕗 When to take:
- Once daily, on an empty stomach, preferably in the morning
- Take with water, at least 30–60 minutes before meals
- ⚠️ Avoid taking with:
- Iron, calcium, soy, antacids (reduce absorption)
- 🔍 Monitoring:
- Check TSH and free T4 levels every 6–8 weeks until levels normalize
- Then monitor every 6–12 months or when clinically indicated
🧾 Adjunct Treatments (Symptom Relief or Associated Conditions):
| 🔹 Condition | 🔹 Management |
|---|---|
| Bradycardia | May require temporary beta-blocker withdrawal |
| Constipation | High-fiber diet, adequate hydration |
| Hyperlipidemia | Statins if persists after euthyroidism |
| Depression | Antidepressants if needed alongside thyroid therapy |
| Anemia | Iron or B12 supplements, based on lab findings |
🚨 Myxedema Coma – Emergency Management:
- ICU admission
- IV levothyroxine and/or liothyronine
- IV corticosteroids (hydrocortisone) to rule out coexisting adrenal insufficiency
- Maintain airway and support vital functions
- Passive warming (if hypothermic)
- Treat underlying cause (infection, trauma, drug overdose)
🛠️ Surgical Management of Hypothyroidism
🔹 Surgery is not a primary treatment for hypothyroidism, but may be considered in specific cases.
🩺 Indications for Surgery:
- Thyroidectomy (Total or Subtotal):
- Thyroid cancer
- Large goiter causing airway compression or dysphagia
- Multinodular goiter or suspicious nodules
- Graves’ disease (as definitive therapy in select cases)
- Post-surgical Outcome:
- Patients will develop permanent hypothyroidism
- Require lifelong levothyroxine therapy
⚠️ Surgical Considerations:
| 🏥 Before Surgery | 🏥 After Surgery |
|---|---|
| Stabilize thyroid levels | Monitor airway, bleeding, voice |
| Control comorbid conditions | Monitor for hypocalcemia (if parathyroids removed) |
| Inform patient about lifelong medication | Watch for signs of thyroid storm in hyperthyroid patients post-op |
🩺 NURSING MANAGEMENT OF HYPOTHYROIDISM
🎯 Goals of Nursing Care:
- Restore and maintain euthyroid state
- Prevent complications (e.g., myxedema coma)
- Promote comfort, activity tolerance, and psychosocial well-being
- Educate patient and family for long-term self-care
🗂️ I. Assessment:
| 🔍 Nursing Assessment Areas | ✅ Key Points |
|---|---|
| Vital Signs | Bradycardia, hypotension, low temperature |
| Skin | Dryness, coolness, pallor, non-pitting edema (myxedema) |
| Cognition and Mood | Depression, slowed responses, memory issues |
| GI Function | Constipation, anorexia |
| Activity Tolerance | Fatigue, muscle weakness |
| Weight and Appetite | Weight gain despite low appetite |
| Menstrual/Reproductive | Irregular periods, infertility |
| Lab Reports | TSH, T3, T4, lipid profile, CBC |
🛏️ II. Nursing Diagnoses (Common Examples):
- Activity intolerance related to fatigue and decreased metabolic rate
- Risk for constipation related to decreased GI motility
- Impaired skin integrity related to dry, thickened skin
- Disturbed thought processes related to reduced cerebral metabolism
- Risk for imbalanced body temperature (hypothermia)
- Knowledge deficit related to lifelong hormone replacement therapy
📝 III. Nursing Interventions:
| 🔧 Intervention | 🩺 Rationale |
|---|---|
| Monitor vital signs regularly | To detect bradycardia, hypotension, or hypothermia |
| Provide a warm, draft-free environment | To manage cold intolerance and prevent hypothermia |
| Allow frequent rest periods | To reduce fatigue and support activity tolerance |
| Encourage high-fiber diet and fluids | To prevent constipation |
| Maintain skin integrity (moisturizers, gentle care) | Dry skin is prone to breakdown |
| Administer prescribed thyroid hormone (Levothyroxine) | To correct hormonal deficiency |
| Monitor for signs of overdose (e.g., palpitations, insomnia) | Indicates possible hyperthyroid state due to over-replacement |
| Educate about lifelong medication adherence | Stopping treatment can cause myxedema coma |
| Teach medication timing (on empty stomach, avoid iron/calcium within 4 hrs) | Improves absorption and efficacy of levothyroxine |
| Provide emotional support and reassurance | Addresses body image changes and depression |
🎓 IV. Patient and Family Education:
- 🔔 Take levothyroxine every morning on an empty stomach
- 🔔 Avoid skipping doses; lifelong therapy is necessary
- 🔔 Avoid taking supplements or antacids near the thyroid medication
- 🔔 Report symptoms of overdose (tachycardia, insomnia, anxiety)
- 🔔 Maintain regular follow-up and TSH monitoring
- 🔔 Educate on hypothyroidism symptoms to detect relapse early
- 🔔 Encourage healthy diet and weight monitoring
🧷 V. Evaluation Criteria (Expected Outcomes):
- Patient maintains stable vital signs within normal limits
- Reports improved energy level and reduced fatigue
- Demonstrates understanding of medication and follow-up
- Shows no signs of myxedema or other complications
- Maintains normal bowel movements and skin integrity
- Participates in self-care and decision-making
⚠️ Complications of Hypothyroidism
If left untreated or poorly managed, hypothyroidism can lead to serious and potentially life-threatening complications.
| 🚨 Complication | 📋 Description |
|---|---|
| Myxedema Coma | Severe, life-threatening form of hypothyroidism. Symptoms include altered mental status, hypothermia, bradycardia, hypotension, respiratory depression. Requires ICU care. |
| Goiter Formation | Enlargement of thyroid gland due to continuous TSH stimulation. Can cause pressure symptoms on trachea/esophagus. |
| Infertility | Disruption of ovulation and menstrual irregularities can lead to difficulty conceiving. |
| Congenital Hypothyroidism (in infants) | If maternal hypothyroidism is untreated during pregnancy → risk of developmental delays, intellectual disability (cretinism). |
| Cardiovascular Issues | Bradycardia, pericardial effusion, increased risk of atherosclerosis, and hyperlipidemia due to altered lipid metabolism. |
| Depression and Cognitive Impairment | Long-standing hypothyroidism may lead to mental sluggishness, memory issues, and depression. |
| Obesity or Weight Gain | Due to reduced metabolic rate. |
| Anemia | Often normocytic or macrocytic due to bone marrow suppression. |
| Sleep Apnea | Secondary to macroglossia and myxedema. |
🧷 Key Points on Hypothyroidism
✅ Definition: Deficiency of thyroid hormones (T3, T4) causing systemic metabolic slowdown.
✅ Common Causes: Hashimoto’s thyroiditis (autoimmune), iodine deficiency, thyroid surgery, radiation, or congenital defects.
✅ Types:
- Primary (thyroid gland issue)
- Secondary (pituitary issue)
- Tertiary (hypothalamus issue)
- Subclinical (mild, asymptomatic)
- Myxedema (severe, life-threatening)
✅ Signs & Symptoms:
- Fatigue, cold intolerance, constipation, dry skin, weight gain, bradycardia, depression, menstrual irregularities.
✅ Diagnosis:
- ↑ TSH, ↓ T3 & T4 (in primary);
- ↓ TSH, ↓ T3 & T4 (in secondary/tertiary);
- Anti-TPO antibodies (in Hashimoto’s)
✅ Treatment:
- Levothyroxine is the treatment of choice
- Regular TSH monitoring is essential
- Lifelong therapy is usually required
✅ Nursing Focus:
- Monitor vitals, prevent complications, educate on medication adherence, and promote self-care.
✅ Complication to watch for:
- Myxedema coma – medical emergency with altered mental status, hypothermia, and organ failure
✅ Patient Education:
- Take medication on an empty stomach
- Avoid drug interactions (e.g., calcium, iron)
- Never stop medication abruptly
- Regular follow-up is essential for dose adjustment
🔥 HYPERTHYROIDISM
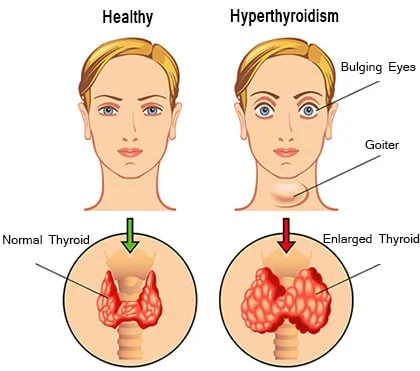
📌 Definition:
Hyperthyroidism is a condition in which the thyroid gland overproduces thyroid hormones — T3 (triiodothyronine) and T4 (thyroxine) — leading to a hypermetabolic state that affects multiple body systems.
🔄 It is the opposite of hypothyroidism and causes an overall increase in body metabolism.
⚠️ Causes of Hyperthyroidism:
| 🔍 Cause | 📋 Description |
|---|---|
| Graves’ Disease (Autoimmune) | Most common cause; body produces TSH receptor antibodies (TRAb) that overstimulate the thyroid |
| Toxic Multinodular Goiter | Presence of multiple autonomously functioning thyroid nodules secreting excess hormone |
| Toxic Adenoma | A single benign tumor (nodule) producing excess thyroid hormone |
| Thyroiditis | Inflammation of the thyroid causing leakage of hormones (e.g., subacute, postpartum thyroiditis) |
| Excessive Iodine Intake | High iodine (e.g., contrast agents, amiodarone) can trigger hormone overproduction in susceptible individuals |
| Overmedication | Taking excess levothyroxine (iatrogenic hyperthyroidism) |
| Pituitary Adenoma | Rare; produces excess TSH, stimulating thyroid (secondary hyperthyroidism) |
| Struma Ovarii | Rare ovarian teratoma that produces thyroid hormone |
🧬 Types of Hyperthyroidism:
| 🏷️ Type | 📖 Description |
|---|---|
| Primary Hyperthyroidism | Due to pathology within the thyroid gland (e.g., Graves’ disease, toxic adenoma) |
| Secondary Hyperthyroidism | Due to increased TSH secretion from the pituitary gland (e.g., TSH-secreting tumor) |
| Tertiary Hyperthyroidism | Due to excess TRH from the hypothalamus (extremely rare) |
| Subclinical Hyperthyroidism | Low TSH, but normal T3/T4; may be asymptomatic or mild |
| Thyroiditis-Induced Hyperthyroidism | Transient hyperthyroidism due to inflammation and hormone leakage (e.g., subacute thyroiditis) |
| Factitious (Iatrogenic) Hyperthyroidism | Due to excess exogenous thyroid hormone intake, often accidental or intentional |
🔬 Pathophysiology of Hyperthyroidism:
- 🧠 Stimulation of the Hypothalamic–Pituitary–Thyroid Axis:
- Normally:
Hypothalamus → TRH → Pituitary → TSH → Thyroid → T3 & T4 - In hyperthyroidism:
Overproduction of T3 and/or T4, often independent of TSH regulation (especially in Graves’ disease or toxic nodules).
- Normally:
- 🔥 Increased Circulating Thyroid Hormones (T3 and T4):
- These hormones increase basal metabolic rate (BMR) and oxygen consumption in tissues.
- ⚙️ Enhanced Metabolic Activity:
- Increased protein breakdown, glucose utilization, lipid metabolism
- Cardiovascular stimulation (↑ heart rate, ↑ cardiac output)
- 🔄 Negative Feedback:
- High T3/T4 levels suppress TSH production via negative feedback, except in TSH-secreting tumors
- 💥 Systemic Overstimulation:
- Multi-organ effects: CNS (anxiety, tremors), CVS (palpitations), GI (diarrhea), Reproductive (amenorrhea), Musculoskeletal (fatigue)
🚨 Signs and Symptoms of Hyperthyroidism:
| 🧠 System | 🔍 Clinical Features |
|---|---|
| 🌡️ General | Weight loss (despite good appetite), heat intolerance, sweating, fatigue |
| 💓 Cardiovascular | Palpitations, tachycardia, hypertension, atrial fibrillation |
| 🧠 Neurological | Nervousness, anxiety, tremors, insomnia, emotional lability |
| 👀 Ocular (Graves’ Disease) | Exophthalmos (bulging eyes), lid lag, gritty sensation |
| 🩺 GI System | Increased bowel movements, diarrhea, hyperdefecation |
| 👩🦰 Skin/Hair | Warm, moist skin; fine hair; thinning hair; flushed face |
| 💃 Musculoskeletal | Muscle weakness, especially proximal muscles (e.g., thighs, shoulders) |
| ♀️ Reproductive | Menstrual irregularities (amenorrhea or oligomenorrhea), infertility |
| 🫁 Respiratory | Shortness of breath, dyspnea on exertion |
| 💤 Others | Sleep disturbances, hyperactivity, restlessness |
🔥 Thyroid Storm (Thyrotoxic Crisis):
A life-threatening emergency with extreme symptoms: high fever, severe tachycardia, altered mental state, and multi-organ failure.
🧪 Diagnosis of Hyperthyroidism:
| 🔬 Test | 📌 Purpose / Interpretation |
|---|---|
| ✅ TSH (Thyroid-Stimulating Hormone) | ↓ Suppressed (low) in primary hyperthyroidism |
| ✅ Free T3 and T4 | ↑ Elevated levels confirm diagnosis |
| ✅ Thyroid Stimulating Immunoglobulins (TSI) | ↑ Positive in Graves’ disease (autoimmune) |
| ✅ Radioactive Iodine Uptake (RAIU) Test | High uptake in Graves’, low in thyroiditis |
| ✅ Thyroid Scan | Identifies hot (functioning) or cold (non-functioning) nodules |
| ✅ Ultrasound of Thyroid | Assesses size, nodules, vascularity |
| ✅ ECG | May show atrial fibrillation, tachycardia |
| ✅ CBC, LFT, Electrolytes | Baseline health and to assess effects of hyperthyroidism or related treatment |
💊 I. MEDICAL MANAGEMENT
🎯 Goals of Medical Treatment:
- Reduce excess thyroid hormone production
- Relieve symptoms
- Prevent complications (e.g., thyroid storm, heart failure)
✅ 1. Antithyroid Medications:
| 💊 Drug | 📋 Description |
|---|---|
| Methimazole (MMI) | First-line antithyroid drug; inhibits thyroid hormone synthesis |
| Propylthiouracil (PTU) | Preferred in pregnancy (1st trimester) and thyroid storm; blocks conversion of T4 → T3 |
| Carbimazole | Prodrug of methimazole (not commonly used in all countries) |
📝 Nursing Tips:
- Monitor for signs of agranulocytosis (sore throat, fever)
- Watch for rash, liver toxicity, GI upset
- Advise regular CBC and LFT checks
✅ 2. Beta-Blockers (Symptom Control):
| 💊 Drug | 📋 Use |
|---|---|
| Propranolol, Atenolol | Control tachycardia, palpitations, tremors, and anxiety caused by excess T3/T4 |
✅ 3. Iodine Therapy:
| 💧 Therapy | 📋 Action |
|---|---|
| Lugol’s iodine or Potassium iodide | Temporarily blocks release of thyroid hormones; used preoperatively or during thyroid storm |
⚠️ Do not use iodine before antithyroid drugs are started, or it may worsen hyperthyroidism.
✅ 4. Radioactive Iodine Therapy (RAI – I-131):
- Most common definitive treatment in adults (non-pregnant)
- Destroys overactive thyroid tissue
- May lead to permanent hypothyroidism requiring lifelong thyroxine therapy
📝 Precautions:
- Not for pregnant/lactating women
- Avoid close contact with others for a few days post-therapy
- Delayed effect (2–4 months), may need interim medications
✅ 5. Corticosteroids:
Used in:
- Thyroid storm
- To reduce TSH receptor antibody activity
- To decrease peripheral conversion of T4 to T3
🛠️ II. SURGICAL MANAGEMENT
🩺 Indications for Thyroid Surgery (Thyroidectomy):
| ⚠️ Indication | 📌 Details |
|---|---|
| Large goiter causing compression | Difficulty breathing/swallowing |
| Suspicious or malignant nodules | Thyroid cancer or suspicious cold nodules |
| Pregnancy | When medications are contraindicated or ineffective |
| Poor compliance with medication/RAI therapy | |
| Severe Graves’ disease not responding to medical therapy |
🔪 Types of Surgery:
| 🩻 Procedure | 📋 Description |
|---|---|
| Subtotal thyroidectomy | Partial removal of thyroid tissue |
| Total thyroidectomy | Complete removal of thyroid gland |
| Lobectomy | Removal of one lobe; done for solitary nodules |
🛏️ Preoperative Nursing Care:
- Stabilize thyroid levels (preferably euthyroid state) with antithyroid drugs
- Administer iodine solution preoperatively (to reduce gland vascularity)
- Monitor vital signs, especially heart rate and BP
- Educate patient on post-op expectations and lifelong hormone therapy (if total removal)
🩺 Postoperative Care:
| 🩹 Focus Area | 🧾 Nursing Action |
|---|---|
| Airway monitoring | Watch for stridor, hoarseness, respiratory distress |
| Bleeding | Monitor dressing, neck swelling, and drain output |
| Calcium monitoring | Risk of hypocalcemia (check for Trousseau’s & Chvostek’s signs) if parathyroids are removed |
| Voice changes | May indicate recurrent laryngeal nerve injury |
| Hormone replacement | Levothyroxine started after total thyroidectomy |
🩺 NURSING MANAGEMENT OF HYPERTHYROIDISM
🎯 Nursing Goals:
- Restore and maintain euthyroid state
- Alleviate signs and symptoms
- Prevent thyroid storm and other complications
- Promote comfort and activity tolerance
- Educate the patient and family about lifelong management and monitoring
🗂️ I. Nursing Assessment
| 🔍 Area | ✅ Assessment Details |
|---|---|
| Vital signs | Tachycardia, hypertension, elevated temperature |
| Neurological status | Restlessness, tremors, anxiety, insomnia |
| GI function | Increased appetite, frequent bowel movements |
| Weight and nutrition | Weight loss despite increased intake |
| Skin and eyes | Warm, moist skin; exophthalmos (in Graves’ disease) |
| Activity level | Fatigue, weakness, intolerance to heat |
| Emotional state | Irritability, mood swings, nervousness |
| Medication history | Use of antithyroid drugs, beta-blockers, previous RAI or surgery |
📝 II. Common Nursing Diagnoses
- Imbalanced nutrition: less than body requirements related to increased metabolic rate
- Activity intolerance related to fatigue and muscle weakness
- Anxiety related to CNS stimulation and disease state
- Risk for decreased cardiac output related to tachyarrhythmias
- Risk for injury (e.g., corneal damage from exophthalmos)
- Disturbed body image related to physical appearance (weight loss, exophthalmos)
🧾 III. Nursing Interventions
| 💡 Intervention | 🩺 Rationale |
|---|---|
| Monitor vital signs frequently | Detect early signs of thyroid storm (↑HR, ↑BP, ↑Temp) |
| Administer medications as prescribed (antithyroid drugs, beta-blockers) | To reduce hormone levels and manage symptoms |
| Provide a cool, calm environment | Minimizes heat intolerance and emotional stress |
| Encourage high-calorie, high-protein diet | Compensates for increased metabolic needs |
| Provide rest periods between activities | Helps manage fatigue |
| Elevate head of bed & protect eyes in Graves’ disease | Reduces periorbital edema and prevents corneal injury |
| Monitor weight and intake/output | To track nutritional and fluid balance |
| Teach stress reduction techniques | Helps avoid triggers for thyroid crisis |
| Prepare patient for surgery or RAI therapy if indicated | Provide pre/post-op education and emotional support |
🧑🏫 IV. Patient and Family Education
- 🔔 Importance of medication adherence (do not stop abruptly)
- 🔔 Signs of overdose or underdose (hyper → hypo symptoms)
- 🔔 Avoid stimulants (e.g., caffeine) and emotional stress
- 🔔 Wear a medical alert bracelet
- 🔔 Regular follow-up for TSH, T3, T4 monitoring
- 🔔 Educate about RAI precautions (if applicable)
- 🔔 Explain lifelong hormone replacement (if post-thyroidectomy)
📊 V. Evaluation Criteria (Expected Outcomes)
- Vital signs within normal limits
- Decreased symptoms of hypermetabolism
- Maintains appropriate nutritional status
- Expresses reduced anxiety and improved emotional well-being
- Demonstrates understanding of the condition and medication regimen
- Prevents complications like thyroid storm or injury
⚠️ Complications of Hyperthyroidism
Uncontrolled or poorly managed hyperthyroidism can lead to serious and potentially life-threatening complications affecting multiple systems:
🔥 1. Thyroid Storm (Thyrotoxic Crisis)
- Medical emergency with sudden exacerbation of symptoms
- Symptoms: High fever (> 40°C), severe tachycardia, hypertension, altered mental status, dehydration, coma
- Triggers: Infection, trauma, surgery, abrupt medication withdrawal
- Requires ICU care, IV antithyroid drugs, beta-blockers, corticosteroids, and cooling measures
💓 2. Cardiac Complications
- Atrial fibrillation, palpitations
- High-output heart failure
- Risk increases in elderly patients and those with preexisting heart disease
🩺 3. Osteoporosis
- Due to increased bone resorption and calcium mobilization
- Especially in long-standing untreated cases
⚠️ 4. Exophthalmos & Eye Damage (Graves’ Ophthalmopathy)
- Can cause corneal ulceration, dryness, double vision
- Severe cases may lead to vision loss
💊 5. Medication Side Effects
- Agranulocytosis (with antithyroid drugs): Watch for fever, sore throat
- Hepatotoxicity: Especially with PTU
- Skin rashes, GI symptoms
🧬 6. Hypothyroidism (Post-Treatment)
- Often occurs after radioactive iodine therapy or thyroidectomy
- Requires lifelong levothyroxine replacement
🧷 Key Points on Hyperthyroidism
✅ Definition: Excess production of thyroid hormones (T3, T4), leading to a hypermetabolic state
✅ Most common cause: Graves’ disease (autoimmune)
✅ Other causes: Toxic multinodular goiter, thyroiditis, thyroid nodules, excessive iodine
✅ Signs & Symptoms:
- Weight loss, heat intolerance, tremors, palpitations, anxiety, diarrhea, exophthalmos
✅ Diagnosis:
- ↓ TSH, ↑ T3/T4
- Positive TSI (in Graves’)
- Radioactive iodine uptake test
✅ Medical Treatment:
- Antithyroid drugs (Methimazole, PTU)
- Beta-blockers for symptom relief
- Radioactive iodine therapy (definitive in adults)
✅ Surgical Treatment:
- Thyroidectomy for large goiters, cancer, or refractory cases
✅ Nursing Focus:
- Monitor vitals, prevent thyroid storm, manage nutrition and rest
- Patient education on medication adherence and follow-up
✅ Complications to Watch:
- Thyroid storm, cardiac arrhythmias, osteoporosis, vision problems
✅ Post-treatment care:
- Monitor for hypothyroidism, teach lifelong hormone replacement if needed
🦋 GOITER
📌 Definition:
A Goiter is an abnormal enlargement of the thyroid gland, which is located in the front of the neck, just below the Adam’s apple.
It may occur with normal, increased, or decreased thyroid function (euthyroid, hyperthyroid, or hypothyroid states).
🗣️ A goiter may or may not be visible but can sometimes cause difficulty in swallowing or breathing if large enough.
⚠️ Causes of Goiter:
| 🔍 Cause Category | 💡 Examples |
|---|---|
| Iodine Deficiency | Most common worldwide cause; leads to decreased hormone production and increased TSH stimulation |
| Autoimmune Thyroid Diseases | Hashimoto’s thyroiditis (hypothyroidism), Graves’ disease (hyperthyroidism) |
| Genetic/Hereditary Factors | Familial goiter tendencies |
| Hormonal Imbalance | During puberty, pregnancy, or menopause |
| Thyroid Nodules | Solitary or multiple nodules may cause thyroid enlargement |
| Inflammation of Thyroid (Thyroiditis) | Subacute, chronic, or silent thyroiditis |
| Overuse of Goitrogens | Foods or drugs that interfere with thyroid hormone synthesis (e.g., cabbage, cassava, amiodarone, lithium) |
| Thyroid Cancer | Can present as a rapidly growing goiter or nodule |
| Radiation Exposure | Previous neck or head radiation may alter thyroid structure |
🧬 Types of Goiter:
✅ A. Based on Function:
| Type | Description |
|---|---|
| Euthyroid Goiter | Normal hormone levels; thyroid is enlarged but functions normally |
| Hypothyroid Goiter | Associated with decreased T3/T4, increased TSH (e.g., Hashimoto’s) |
| Hyperthyroid Goiter | Associated with increased T3/T4 and suppressed TSH (e.g., Graves’ disease) |
✅ B. Based on Morphology (Appearance):
| Type | Description |
|---|---|
| Diffuse Goiter | Uniformly enlarged thyroid without nodules; seen in early iodine deficiency or Graves’ |
| Nodular Goiter | Thyroid gland has one or more lumps or nodules |
| → Uninodular (Solitary Nodule) | Single nodule causing enlargement |
| → Multinodular Goiter (MNG) | Multiple nodules causing an irregular, bumpy gland |
| Retrosternal Goiter | Enlarged thyroid extends behind the sternum; may compress trachea or esophagus |
| Toxic Goiter | Produces excess hormones (seen in Graves’ or toxic nodules) |
| Nontoxic Goiter | Enlarged gland without hormone overproduction (usually euthyroid or hypothyroid) |
🧬 Pathophysiology of Goiter:
- ⚖️ Imbalance in Thyroid Hormone Production:
- In iodine deficiency or autoimmune conditions, the thyroid cannot produce enough T3 and T4.
- The pituitary gland responds by releasing more TSH (thyroid-stimulating hormone).
- 🔄 TSH Overstimulation:
- Excess TSH stimulates the thyroid follicles, causing hyperplasia and hypertrophy of thyroid cells.
- 🌱 Thyroid Gland Enlargement:
- Leads to diffuse or nodular goiter formation.
- In some cases, nodules may become autonomous (function independently of TSH), producing excess hormone → toxic goiter.
- ⚠️ Other Causes:
- In Graves’ disease, autoantibodies (TSI) mimic TSH, stimulating uncontrolled thyroid growth and hormone production.
- In Hashimoto’s thyroiditis, chronic inflammation causes destruction and regeneration of thyroid tissue, leading to goiter.
🚨 Signs and Symptoms of Goiter:
🗣️ Symptoms depend on size, location, and functionality (hypo-, hyper-, or euthyroid)
✅ Local Symptoms (Due to Size/Compression):
| 🔍 Area | 🚨 Signs & Symptoms |
|---|---|
| Neck | Visible swelling in the front of the neck (may move when swallowing) |
| Swallowing | Dysphagia (difficulty swallowing), especially with large goiters |
| Breathing | Dyspnea, stridor, especially if retrosternal or compressing trachea |
| Voice | Hoarseness (due to recurrent laryngeal nerve compression) |
✅ Systemic Symptoms (Based on Thyroid Function):
🧊 If Hypothyroid (e.g., Hashimoto’s):
- Fatigue
- Weight gain
- Cold intolerance
- Dry skin, constipation
- Depression
🔥 If Hyperthyroid (e.g., Graves’):
- Weight loss
- Palpitations
- Heat intolerance
- Nervousness, tremors
- Diarrhea, insomnia
😐 If Euthyroid:
- Usually asymptomatic, except for visible or palpable neck mass
🧪 Diagnosis of Goiter:
| 🧬 Test | 📌 Purpose / Interpretation |
|---|---|
| ✅ Thyroid Function Tests (TFTs) | TSH, Free T3, Free T4 → Determines if hypo-, hyper-, or euthyroid |
| ✅ Anti-TPO Antibodies | Positive in Hashimoto’s thyroiditis |
| ✅ TSI (Thyroid Stimulating Immunoglobulins) | Elevated in Graves’ disease |
| ✅ Neck Ultrasound | Assesses thyroid size, nodules, cystic vs solid areas |
| ✅ Fine Needle Aspiration (FNA) | For biopsy of suspicious nodules (to rule out malignancy) |
| ✅ Radioactive Iodine Uptake (RAIU) Scan | Differentiates between toxic (hot) nodules and non-functioning (cold) nodules |
| ✅ X-ray / CT Scan (Neck/Chest) | To assess tracheal deviation, compression, or retrosternal extension |
💊 I. MEDICAL MANAGEMENT
🎯 Goals:
- Control the underlying cause
- Normalize thyroid hormone levels
- Reduce the size of the goiter
- Relieve compression symptoms (if present)
✅ 1. Iodine Supplementation
- Used in iodine-deficiency-related goiters
- Oral potassium iodide or iodized salt in endemic areas
- Not effective in nodular or autoimmune goiters
- ⚠️ Excess iodine may worsen autoimmune thyroid disorders
✅ 2. Thyroid Hormone Replacement Therapy (Levothyroxine)
- Used for goiter associated with hypothyroidism (e.g., Hashimoto’s)
- Suppressive therapy may reduce TSH stimulation and shrink the goiter
- Dosage adjusted to maintain normal TSH levels
- Requires lifelong treatment in many cases
✅ 3. Antithyroid Drugs (e.g., Methimazole, PTU)
- Used in toxic goiters (hyperthyroidism)
- Suppresses overproduction of thyroid hormones
- Used short- or long-term depending on severity
✅ 4. Beta-Blockers (e.g., Propranolol)
- Used to control symptoms of hyperthyroidism (palpitations, tremors)
- Not a definitive treatment but provides symptomatic relief
✅ 5. Radioactive Iodine Therapy (RAI – I-131)
- Used for toxic multinodular goiter or Graves’ disease
- Destroys overactive thyroid tissue
- Often leads to hypothyroidism, requiring levothyroxine replacement
- Not suitable for pregnant/lactating women or patients with severe compressive symptoms
🛠️ II. SURGICAL MANAGEMENT
🎯 Goals:
- Remove enlarged thyroid tissue causing compression, cosmetic deformity, or suspicious nodules/cancer
🔍 Indications for Surgery (Thyroidectomy):
| ⚠️ Indication | 📌 Details |
|---|---|
| Large goiter causing compression | Dysphagia, dyspnea, stridor |
| Suspicious/malignant nodules | Cold nodule or confirmed thyroid cancer |
| Retrosternal (substernal) goiter | Extension into chest cavity |
| Toxic multinodular goiter or toxic adenoma | Unresponsive to medical therapy |
| Cosmetic reasons | For visibly disfiguring neck swelling |
| Non-responsive to RAI or medication | Persistent or recurrent symptoms |
✂️ Types of Thyroid Surgery:
| 🩻 Procedure | 📋 Description |
|---|---|
| Lobectomy | Removal of one lobe; for solitary benign nodule |
| Subtotal Thyroidectomy | Partial removal of both lobes; leaves some thyroid tissue |
| Total Thyroidectomy | Complete removal; used in cancer or diffuse toxic goiter |
| Isthmusectomy | Removal of the isthmus (central portion); for small nodules limited to the isthmus |
🛏️ Preoperative Care:
- Achieve euthyroid state (in hyperthyroid patients) with medications
- Administer iodine solution pre-op (to reduce vascularity)
- Explain the procedure, risks (nerve damage, hypocalcemia), and post-op care
- Baseline vital signs, calcium levels, and airway assessment
🩺 Postoperative Care:
| 🎯 Focus Area | 🩹 Nursing Action |
|---|---|
| Airway Management | Watch for stridor, hoarseness, respiratory distress (due to hematoma or laryngeal nerve injury) |
| Bleeding | Inspect surgical site and dressing regularly |
| Calcium Monitoring | Watch for hypocalcemia → Trousseau’s and Chvostek’s signs |
| Voice Monitoring | Check for hoarseness (possible recurrent laryngeal nerve injury) |
| Thyroid Hormone Replacement | Begin levothyroxine after total thyroidectomy |
| Pain Control | Administer analgesics and encourage soft neck movements |
🩺 NURSING MANAGEMENT OF GOITER
🎯 Nursing Goals:
- Relieve symptoms
- Monitor and manage thyroid function
- Prevent complications (e.g., airway obstruction, hypothyroidism)
- Prepare and support patient through medical or surgical treatment
- Educate patient on condition, medication, and self-care
🗂️ I. Nursing Assessment
| 🔍 Area | ✅ Key Focus |
|---|---|
| Neck Examination | Observe for visible swelling, symmetry, movement with swallowing |
| Airway and Breathing | Check for stridor, hoarseness, or dyspnea (especially with large or retrosternal goiter) |
| Swallowing | Assess for dysphagia or pressure on esophagus |
| Thyroid Function Symptoms | Signs of hypo-/hyperthyroidism (weight change, fatigue, palpitations, heat/cold intolerance) |
| Voice Changes | Monitor for hoarseness (indicates laryngeal nerve involvement) |
| Lab Reports | TSH, Free T3, Free T4, thyroid antibodies, calcium levels (pre/post-op) |
📝 II. Common Nursing Diagnoses
- Ineffective airway clearance related to tracheal compression
- Imbalanced nutrition related to altered metabolism (hyper- or hypothyroid state)
- Risk for aspiration related to dysphagia
- Risk for impaired verbal communication related to laryngeal nerve damage
- Deficient knowledge related to disease process, medications, and surgical care
- Anxiety related to visible neck swelling, diagnostic procedures, or surgery
🧾 III. Nursing Interventions
🔹 Monitoring and Symptom Management
| 💡 Intervention | 🩺 Rationale |
|---|---|
| Monitor vital signs (esp. HR, BP, temp) | Detect hyperthyroid or hypothyroid state |
| Assess for signs of airway obstruction | Large goiter or retrosternal extension can compress trachea |
| Observe for voice changes or hoarseness | May indicate nerve compression or surgical injury |
| Administer prescribed medications | Antithyroid drugs, levothyroxine, beta-blockers |
| Monitor lab values regularly | TSH, T3, T4 to guide medication and treatment decisions |
🔹 Postoperative Care (If Thyroidectomy Done)
| 🩺 Intervention | 🔎 Purpose |
|---|---|
| Elevate head of bed | Reduces neck swelling and promotes airway drainage |
| Monitor for bleeding at incision site | Early sign of hematoma or surgical complication |
| Monitor calcium levels | Hypocalcemia may result from parathyroid injury |
| Assess for Trousseau’s and Chvostek’s signs | Early signs of hypocalcemia |
| Support neck when moving or coughing | Prevents strain on the surgical site |
| Provide pain relief and wound care | Promotes comfort and healing |
🔹 Patient and Family Education
- 💬 Explain the cause and type of goiter (e.g., iodine deficiency, autoimmune)
- 💊 Emphasize medication adherence (antithyroid or thyroid hormone replacement)
- 🧂 Teach about iodine-rich foods (seafood, dairy, iodized salt) if iodine deficiency is the cause
- 📅 Encourage regular follow-up and blood testing
- 🎓 Educate about signs of hypo- and hyperthyroidism
- 🩺 Post-thyroidectomy: explain need for lifelong levothyroxine (if total removal)
📊 IV. Evaluation Criteria (Expected Outcomes)
- Patient maintains clear airway and normal breathing
- Reports relief from pressure symptoms (dysphagia, hoarseness)
- Maintains stable thyroid hormone levels (T3, T4, TSH)
- Demonstrates understanding of medication and follow-up needs
- Prevents surgical complications (hypocalcemia, hemorrhage)
- Expresses reduced anxiety and improved quality of life
⚠️ COMPLICATIONS OF GOITER
If a goiter is left untreated or poorly managed, especially when large or toxic, it can lead to several local, systemic, and endocrine-related complications:
🧨 1. Compressive Complications
| 🚨 Complication | 📋 Description |
|---|---|
| Tracheal compression | Causes dyspnea, stridor, or airway obstruction |
| Esophageal compression | Leads to dysphagia (difficulty swallowing) |
| Recurrent laryngeal nerve compression | Hoarseness or voice changes |
| Superior vena cava syndrome | Rare; large retrosternal goiters may compress great vessels |
🔥 2. Thyroid Functional Complications
| 🔥 Type | 🔍 Description |
|---|---|
| Hypothyroidism | In long-standing or autoimmune goiters (e.g., Hashimoto’s) |
| Hyperthyroidism (Toxic Goiter) | Seen in Graves’ disease, toxic multinodular goiter |
| Thyroid storm | Life-threatening complication of toxic goiter if unmanaged |
🧬 3. Malignant Transformation
- Some cold nodules in multinodular goiter may be malignant
- Requires evaluation with FNAC or biopsy
🧪 4. Postoperative Complications (Thyroidectomy)
| ⚠️ Complication | 📌 Description |
|---|---|
| Hypocalcemia | Due to accidental removal/injury of parathyroid glands |
| Hemorrhage/hematoma | Can cause airway obstruction |
| Infection | Surgical site infection |
| Voice changes | Damage to recurrent laryngeal nerve |
🧷 KEY POINTS ON GOITER
✅ Definition: Enlargement of the thyroid gland, which may be diffuse or nodular, and functional (toxic) or non-functional (nontoxic)
✅ Causes: Iodine deficiency, autoimmune diseases (Graves’, Hashimoto’s), thyroid nodules, goitrogens, inflammation, tumors
✅ Types:
- Based on function: Euthyroid, hypothyroid, hyperthyroid
- Based on morphology: Diffuse, nodular (uninodular or multinodular), retrosternal, toxic or nontoxic
✅ Symptoms: Neck swelling, dysphagia, dyspnea, voice changes, or symptoms of thyroid dysfunction
✅ Diagnosis: TFTs (TSH, T3, T4), ultrasound, RAIU scan, FNAC, antibody testing
✅ Treatment:
- Medical: Levothyroxine, antithyroid drugs, iodine supplementation, RAI therapy
- Surgical: Thyroidectomy for compressive symptoms, cosmetic concerns, malignancy, or failure of medical therapy
✅ Nursing Role: Monitor airway, manage symptoms, educate patient, post-op care, promote adherence
✅ Complications: Compression of nearby structures, thyroid dysfunction, malignancy, and post-surgical complications.
🧬 THYROIDITIS
📌 Definition:
Thyroiditis is a general term for inflammation of the thyroid gland, which may be acute, subacute, or chronic in nature. It may result in hypothyroidism, hyperthyroidism, or transient thyroid dysfunction, depending on the type and stage of inflammation.
⚠️ The inflammation may be infectious, autoimmune, post-viral, drug-induced, or radiation-related.
⚠️ Causes of Thyroiditis:
| 🔍 Cause Category | 📋 Examples |
|---|---|
| Autoimmune | Hashimoto’s thyroiditis, postpartum thyroiditis |
| Viral (post-viral) | Subacute (De Quervain’s) thyroiditis after upper respiratory infections |
| Bacterial (infectious) | Acute suppurative thyroiditis from bacterial invasion |
| Drugs | Amiodarone, interferon-alpha, lithium |
| Radiation-induced | After radioactive iodine therapy or external beam radiation |
| Trauma or surgery | Injury to the thyroid gland |
| Postpartum hormonal changes | Postpartum thyroiditis due to immune reactivation |
| Genetic | Certain HLA types predispose to autoimmune thyroiditis |
🧬 Types of Thyroiditis:
🔹 1. Hashimoto’s Thyroiditis (Chronic Lymphocytic Thyroiditis)
- Most common type, especially in women
- Autoimmune destruction of thyroid gland
- Gradual development of hypothyroidism
- Associated with anti-TPO and anti-Tg antibodies
🔹 2. Subacute Thyroiditis (De Quervain’s Thyroiditis)
- Post-viral inflammation (e.g., after mumps, influenza, adenovirus)
- Painful, tender, swollen thyroid
- Initially causes hyperthyroidism, followed by hypothyroidism, then recovery
- Self-limiting (usually resolves in weeks to months)
🔹 3. Acute (Suppurative) Thyroiditis
- Bacterial infection of the thyroid (rare but serious)
- High fever, severe neck pain, and redness
- May form an abscess
- Requires antibiotics and sometimes surgical drainage
🔹 4. Silent (Painless) Thyroiditis
- Autoimmune, similar to Hashimoto’s but transient
- Painless, mild thyroid enlargement
- Often seen postpartum (postpartum thyroiditis)
- May present with transient hyperthyroidism, followed by hypothyroidism
🔹 5. Postpartum Thyroiditis
- Occurs within 1 year of delivery
- Autoimmune in nature
- Often follows a course of hyperthyroid → hypothyroid → recovery
🔹 6. Drug-Induced Thyroiditis
- Caused by medications like:
- Amiodarone
- Interferon-alpha
- Lithium
- Can cause either hypothyroidism or hyperthyroidism
🔹 7. Radiation-Induced Thyroiditis
- Occurs after radioactive iodine therapy (RAI) or radiation to neck area
- May cause transient hyperthyroidism due to release of stored hormones
🔁 Pathophysiology of Thyroiditis:
The underlying pathophysiology varies depending on the type of thyroiditis, but the general process involves:
- Triggering Event (Infection, Autoimmune, Drug):
- A viral infection, autoimmune reaction, or drug exposure leads to inflammation of the thyroid gland.
- Thyroid Cell Damage:
- Inflammation causes destruction of thyroid follicular cells, leading to release of stored thyroid hormones (T3, T4) into the bloodstream.
- Transient Hyperthyroidism:
- This sudden release leads to temporary hyperthyroidism (thyrotoxic phase), typically lasting weeks.
- Hormone Depletion:
- As the hormone stores are depleted and gland is unable to produce more due to damage, hypothyroidism may follow.
- Resolution or Progression:
- In most cases (e.g., subacute, postpartum), thyroid function returns to normal (euthyroid).
- In Hashimoto’s, the autoimmune destruction progresses to permanent hypothyroidism.
🚨 Signs and Symptoms of Thyroiditis (Based on Phase and Type):
🟠 General Symptoms (Common to Many Types):
| 🧠 System | 🔍 Symptoms |
|---|---|
| Neck/Local | Pain (subacute, acute), swelling, tenderness, warmth |
| General | Fatigue, malaise, weight change, fever (acute) |
🔥 Hyperthyroid Phase (Thyrotoxic Phase):
Seen in early stages of subacute, silent, postpartum thyroiditis
- Palpitations
- Heat intolerance
- Weight loss
- Nervousness, anxiety
- Tremors
- Sweating
- Increased bowel movements
❄️ Hypothyroid Phase:
Seen in later stages or in Hashimoto’s and postpartum thyroiditis
- Fatigue
- Weight gain
- Cold intolerance
- Dry skin, hair loss
- Constipation
- Depression
- Menstrual irregularities
⚠️ Type-Specific Symptoms:
| Type | Distinct Symptoms |
|---|---|
| Subacute (De Quervain’s) | Painful, tender, enlarged thyroid; follows URI |
| Acute Suppurative | High fever, neck redness, pus, dysphagia |
| Hashimoto’s | Painless goiter, gradual fatigue, common in women |
| Postpartum | Occurs within 1 year of delivery, painless thyroid swelling |
| Silent thyroiditis | Mild thyrotoxic symptoms without pain |
🧪 Diagnosis of Thyroiditis:
| 🧬 Test | 📌 Interpretation |
|---|---|
| ✅ Thyroid Function Tests (TFTs) |
- Hyperthyroid phase: ↓ TSH, ↑ T3/T4
- Hypothyroid phase: ↑ TSH, ↓ T3/T4
- Euthyroid: Normal values |
| ✅ Thyroid Antibodies |
- Anti-TPO and anti-Tg: Positive in Hashimoto’s and postpartum thyroiditis
- TSI (Thyroid Stimulating Immunoglobulin): Usually negative in thyroiditis (positive in Graves’ disease) |
| ✅ ESR (Erythrocyte Sedimentation Rate) |
- Elevated in subacute thyroiditis (indicates inflammation) |
| ✅ CRP (C-Reactive Protein) |
- Elevated in acute and subacute thyroiditis |
| ✅ Thyroid Ultrasound |
- Diffuse heterogeneity in Hashimoto’s
- Hypoechoic areas in subacute or silent types
- Abscess in acute suppurative thyroiditis |
| ✅ Radioactive Iodine Uptake (RAIU) Scan |
- Decreased uptake in thyroiditis (due to hormone leakage)
- Helps distinguish from Graves’ disease (which has high uptake) |
| ✅ Fine Needle Aspiration (FNA) |
- Used in acute thyroiditis to rule out abscess or malignancy |
💊 I. MEDICAL MANAGEMENT
🎯 Goals of Medical Management:
- Relieve symptoms (pain, swelling, hormonal imbalance)
- Treat the underlying cause (e.g., infection, autoimmune)
- Prevent complications such as hypothyroidism or abscess formation
🔹 1. Subacute Thyroiditis (De Quervain’s):
| 🧾 Medication | 💡 Purpose |
|---|---|
| NSAIDs (e.g., Ibuprofen) | First-line for pain and inflammation |
| Corticosteroids (e.g., Prednisone) | For severe pain or NSAID-resistant cases |
| Beta-blockers (e.g., Propranolol) | Control hyperthyroid symptoms (palpitations, tremors) during thyrotoxic phase |
| Levothyroxine | Temporary, if patient develops hypothyroid phase |
📌 Usually self-limiting and resolves within weeks to months
🔹 2. Hashimoto’s Thyroiditis:
| 🧾 Medication | 💡 Purpose |
|---|---|
| Levothyroxine (T4 hormone replacement) | Mainstay treatment for permanent hypothyroidism |
| No antithyroid drugs are used | As hyperthyroidism is due to hormone leakage, not overproduction |
🔹 3. Silent & Postpartum Thyroiditis:
- Beta-blockers for thyrotoxic phase
- Levothyroxine for temporary or long-term hypothyroid phase
- Monitor thyroid function every 6–8 weeks, as many cases resolve spontaneously
🔹 4. Acute (Suppurative) Thyroiditis:
| 🧾 Treatment | 💡 Purpose |
|---|---|
| Broad-spectrum antibiotics | Treat underlying bacterial infection |
| IV fluids & supportive care | If systemic infection/sepsis suspected |
| Drainage of abscess (if present) | May require surgical drainage |
📌 This type is rare but a medical emergency
🔹 5. Drug-induced Thyroiditis:
- Discontinue offending drug (e.g., amiodarone, lithium)
- Treat symptoms based on phase (hyper/hypothyroid)
- Monitor function, may normalize after drug cessation
🛠️ II. SURGICAL MANAGEMENT
Surgery is rarely required in thyroiditis but may be considered in select situations.
🔪 Indications for Surgery (Thyroidectomy):
| ⚠️ Indication | 📋 Description |
|---|---|
| Persistent large goiter | Causing compressive symptoms (e.g., dysphagia, dyspnea) |
| Suspicion of malignancy | Focal nodules or indeterminate FNAC results in Hashimoto’s |
| Recurrent painful thyroiditis | Uncommon, but may be seen in subacute cases |
| Abscess formation (acute) | May need surgical drainage if not resolved by aspiration |
🩺 Surgical Procedures:
| 🛠️ Procedure | 📌 Use |
|---|---|
| Lobectomy | Removal of one lobe with localized pathology |
| Total thyroidectomy | In diffuse or bilateral involvement, or suspected malignancy |
| Incision and drainage | For abscess in acute suppurative thyroiditis |
🛏️ Pre- and Post-Operative Considerations:
- Ensure thyroid function is stable (euthyroid) before elective surgery
- Monitor for bleeding, infection, hypocalcemia, and voice changes post-op
- Educate patient on lifelong hormone therapy if total thyroidectomy is performed
🩺 NURSING MANAGEMENT OF THYROIDITIS
🎯 Goals of Nursing Care:
- Alleviate symptoms (pain, swelling, fatigue)
- Prevent complications (e.g., hypothyroidism, thyroid storm, airway compromise)
- Support medical/surgical interventions
- Promote patient education and long-term monitoring
- Encourage emotional and psychological well-being
🗂️ I. Nursing Assessment
| 🔍 Area | ✅ Key Focus |
|---|---|
| Vital signs | Monitor temperature, pulse, BP—especially in thyrotoxic or septic patients |
| Thyroid gland | Assess for swelling, tenderness, warmth, asymmetry, or hardness |
| Swallowing and voice | Check for dysphagia, hoarseness, or voice changes |
| Thyroid symptoms | Look for hyperthyroid signs (anxiety, tremors, heat intolerance) and hypothyroid signs (fatigue, cold intolerance, weight gain) |
| Pain assessment | Especially important in subacute and acute types |
| Lab reports | Review thyroid function tests (TSH, T3, T4), antibody levels, ESR, CRP, WBC count |
📝 II. Common Nursing Diagnoses
- Acute pain related to thyroid inflammation (subacute or acute thyroiditis)
- Risk for ineffective airway clearance related to swelling or abscess formation
- Activity intolerance related to hormonal imbalance (hypo-/hyperthyroidism)
- Deficient knowledge regarding disease process and treatment plan
- Risk for infection (in suppurative thyroiditis or post-surgery)
- Imbalanced nutrition related to metabolic dysfunction
🧾 III. Nursing Interventions
🔹 For Subacute or Hashimoto’s Thyroiditis:
| 💡 Intervention | 🩺 Rationale |
|---|---|
| Provide warm compresses to neck (if ordered) | Reduces discomfort in subacute thyroiditis |
| Administer NSAIDs or corticosteroids as prescribed | Relieves inflammation and pain |
| Monitor for signs of hypothyroidism | Important as disease often progresses to low hormone states |
| Educate about need for regular TSH monitoring | To track function and adjust levothyroxine if needed |
| Encourage rest during fatigue phases | Helps manage energy levels |
🔹 For Acute (Suppurative) Thyroiditis:
| 💡 Intervention | 🩺 Rationale |
|---|---|
| Administer antibiotics as prescribed | Treats underlying bacterial infection |
| Monitor for fever, swelling, redness | Early signs of abscess or worsening infection |
| Prepare for abscess drainage if required | Prevents airway compromise or spread of infection |
| Maintain airway and monitor respiratory effort | Goiter or swelling may compress airway |
🔹 For Thyrotoxic Phase (Silent/Postpartum Thyroiditis):
| 💡 Intervention | 🩺 Rationale |
|---|---|
| Administer beta-blockers (e.g., propranolol) as ordered | Controls symptoms like palpitations, tremors |
| Monitor vital signs closely (especially HR and BP) | Detects early signs of thyrotoxic crisis |
| Educate patient on symptoms of worsening hyperthyroidism | Ensures timely reporting and intervention |
🔹 Post-Surgical Nursing Care (if thyroidectomy is done):
| 🩺 Focus Area | ✅ Action |
|---|---|
| Airway assessment | Watch for stridor, hoarseness, respiratory distress |
| Incision care | Check for bleeding, swelling, signs of infection |
| Voice monitoring | Evaluate for recurrent laryngeal nerve damage |
| Calcium monitoring | Check for signs of hypocalcemia (Trousseau’s, Chvostek’s signs) |
| Hormone replacement | Educate on lifelong levothyroxine if total thyroidectomy is done |
🧑🏫 IV. Patient and Family Education
- Explain the type and nature of thyroiditis (autoimmune, viral, bacterial)
- Stress the importance of regular thyroid function tests
- Teach correct medication usage (e.g., levothyroxine, steroids, NSAIDs)
- Warn about symptoms of hypo- or hyperthyroidism
- Reassure that many forms (e.g., subacute, postpartum) are self-limiting
- Discuss importance of follow-up for long-term monitoring
📊 V. Evaluation Criteria (Expected Outcomes)
- Pain is reduced or controlled
- Thyroid hormone levels return to normal range
- No signs of airway obstruction or infection
- Patient demonstrates understanding of disease and medication regimen
- Patient participates actively in follow-up and self-monitoring
⚠️ COMPLICATIONS OF THYROIDITIS
Complications of thyroiditis depend on the type, severity, and duration of the condition. If left untreated or poorly managed, thyroiditis can lead to significant health issues.
🔥 1. Hypothyroidism (Most Common)
- Especially in Hashimoto’s thyroiditis
- May be permanent, requiring lifelong levothyroxine therapy
- Occurs after destruction of thyroid follicles
⚠️ 2. Hyperthyroidism (Thyrotoxic Phase)
- Seen in early stages of subacute, silent, and postpartum thyroiditis
- Can cause cardiac complications like palpitations, atrial fibrillation, and heart failure if severe
😷 3. Thyroid Storm (Rare)
- A medical emergency seen in uncontrolled thyrotoxic phase
- High fever, tachycardia, altered mental status
- Requires ICU management
🦠 4. Abscess Formation and Sepsis
- In acute suppurative thyroiditis (bacterial origin)
- Risk of airway obstruction, neck cellulitis, or septicemia
🩺 5. Compressive Symptoms
- Large or inflamed gland may compress the trachea or esophagus, causing:
- Dysphagia (difficulty swallowing)
- Dyspnea (difficulty breathing)
- Hoarseness due to recurrent laryngeal nerve involvement
🔬 6. Malignancy Risk (Rare)
- Long-standing Hashimoto’s thyroiditis slightly increases the risk of thyroid lymphoma or papillary thyroid carcinoma
🧷 KEY POINTS ON THYROIDITIS
✅ Definition: Inflammation of the thyroid gland, which may be autoimmune, viral, bacterial, or drug-induced
✅ Common Types:
- Hashimoto’s (chronic, autoimmune, leads to hypothyroidism)
- Subacute (post-viral, painful, self-limiting)
- Silent and Postpartum (painless, transient thyroid dysfunction)
- Acute Suppurative (bacterial, painful, requires urgent care)
✅ Phases: Many types follow a pattern →
Thyrotoxic phase → Hypothyroid phase → Recovery
✅ Diagnosis:
- TFTs (TSH, T3, T4)
- Anti-TPO antibodies (Hashimoto’s)
- ESR/CRP (inflammatory markers)
- Ultrasound, RAIU, FNAC (if nodular or suspicious)
✅ Medical Treatment:
- NSAIDs, steroids for inflammation
- Levothyroxine for hypothyroidism
- Antibiotics for acute bacterial thyroiditis
- Beta-blockers for symptom control in hyperthyroidism
✅ Surgery: Rare, used in abscess, compressive goiter, or suspicion of malignancy
✅ Nursing Role:
- Monitor thyroid function
- Manage symptoms (pain, fever, hormonal imbalance)
- Provide patient education
- Prevent and detect complications early
🧬 Thyroid Cysts and Tumors
📌 Definition:
- Thyroid cysts are fluid-filled sacs that develop within the thyroid gland. Most are benign and part of nodular thyroid disease.
- Thyroid tumors are solid or mixed (solid + cystic) growths in the thyroid, which can be benign (non-cancerous) or malignant (cancerous).
⚠️ Causes and Risk Factors:
| 🔍 Cause | 📋 Description |
|---|---|
| Iodine deficiency | Leads to nodular goiter formation, which can develop cysts/tumors |
| Genetic mutations | Mutations in genes like RET, BRAF, RAS (esp. in thyroid cancer) |
| Radiation exposure | Especially in childhood (neck radiation increases cancer risk) |
| Chronic thyroiditis | Long-standing Hashimoto’s may increase cancer risk |
| Hormonal imbalance | May influence cyst formation |
| Family history | Thyroid cancer or MEN syndromes |
🧬 Types of Thyroid Cysts and Tumors:
🔹 1. Thyroid Cysts:
- Simple cysts: Benign, purely fluid-filled
- Complex cysts: Partially solid, may need further evaluation
- Colloid cysts: Associated with nodular goiter; usually benign
🔹 2. Benign Thyroid Tumors:
- Follicular adenoma: Common, encapsulated, and non-invasive
- Hurthle cell adenoma: Variant of follicular adenoma, may mimic malignancy
🔹 3. Malignant Thyroid Tumors (Thyroid Cancer):
| 🔬 Type | 📋 Description |
|---|---|
| Papillary carcinoma | Most common (80–85%), slow-growing, good prognosis |
| Follicular carcinoma | Moderate prognosis; may spread via blood |
| Medullary carcinoma | Arises from parafollicular (C) cells; secretes calcitonin |
| Anaplastic carcinoma | Rare, aggressive, poor prognosis |
| Thyroid lymphoma | Rare; often linked to Hashimoto’s thyroiditis |
🔁 Pathophysiology:
- Cellular mutation or hyperplasia occurs in thyroid follicular cells
- Leads to abnormal cell proliferation → forming nodules or tumors
- Cyst formation may occur from degeneration or hemorrhage within nodules
- Malignant transformation may occur due to genetic or environmental triggers
- Some tumors (e.g., medullary) produce hormones or peptides, causing systemic effects
🚨 Signs and Symptoms:
| 🧠 System | 🔍 Clinical Signs |
|---|---|
| Local/Neck | Painless neck swelling or lump, visible or palpable nodule |
| Swallowing/Breathing | Dysphagia, hoarseness, stridor (if compressive) |
| Thyroid dysfunction | Usually euthyroid, but may have hyper/hypothyroid signs |
| Malignancy indicators | Rapid growth, hard consistency, fixed mass, lymphadenopathy |
| Medullary carcinoma | May cause diarrhea or flushing (due to calcitonin secretion) |
🧪 Diagnosis:
| 🔬 Test | 📋 Purpose |
|---|---|
| ✅ Thyroid Function Tests (TFTs) | TSH, T3, T4 — to assess gland function |
| ✅ Neck Ultrasound | To evaluate nodule size, structure, solid/cystic nature |
| ✅ Fine Needle Aspiration (FNA) Biopsy | Gold standard for differentiating benign vs malignant |
| ✅ Thyroid Scan (RAIU) | Hot (functional) vs Cold (non-functional) nodules |
| ✅ Serum Calcitonin | Elevated in medullary carcinoma |
| ✅ Thyroglobulin levels | Used in cancer follow-up |
| ✅ CT/MRI of neck | To assess retrosternal extension or lymph node involvement |
💊 Medical Management:
| 💊 Treatment | 📋 Use |
|---|---|
| Levothyroxine therapy | TSH suppression in benign nodular goiter or post-op cancer |
| Ethanol injection | Minimally invasive treatment for benign cysts |
| Radioactive iodine therapy (RAI) | For papillary/follicular cancer post-surgery |
| Targeted therapy (e.g., TKIs) | For advanced or metastatic thyroid cancers |
| Chemotherapy | Used only in aggressive tumors (e.g., anaplastic carcinoma) |
| Observation | For small, stable, benign nodules or cysts |
🛠️ Surgical Management:
| ✂️ Surgery Type | 📋 Indications |
|---|---|
| Lobectomy (hemithyroidectomy) | Solitary benign nodule, diagnostic uncertainty |
| Total thyroidectomy | Thyroid cancer, bilateral nodular disease, large goiters |
| Near-total thyroidectomy | Cancer with minimal tissue left to protect parathyroids |
| Lymph node dissection | If metastasis to cervical nodes is present |
| Drainage | For symptomatic or infected thyroid cysts |
🩺 Nursing Management:
🔹 Preoperative Care:
- Monitor thyroid hormone levels, ECG (if hyperthyroid)
- Educate about procedure, anesthesia, post-op expectations
- Administer pre-op iodine (if ordered) to reduce gland vascularity
🔹 Postoperative Care:
| Nursing Focus | Actions |
|---|---|
| Airway management | Monitor for stridor, dyspnea (due to hematoma or laryngeal edema) |
| Bleeding | Check surgical site, dressing, drain output |
| Calcium monitoring | Assess for hypocalcemia (Trousseau’s/Chvostek’s signs) |
| Voice monitoring | Detect recurrent laryngeal nerve damage |
| Hormone replacement | Start lifelong levothyroxine if total thyroidectomy done |
| Pain management and wound care | Provide analgesia and maintain sterile dressing |
🔹 Patient Education:
- Teach about TSH monitoring, signs of hypo-/hyperthyroidism
- Emphasize medication adherence
- Inform about follow-up ultrasound/FNA if nodule is retained
⚠️ Complications:
| 🚨 Type | Examples |
|---|---|
| Local | Hematoma, infection, vocal cord paralysis |
| Systemic | Hypothyroidism, hypocalcemia, recurrence |
| Cancer-related | Metastasis (lungs, bones), recurrence, airway invasion |
| Post-op | Thyroid storm (rare), voice changes, permanent hormone dependence |
🧷 Key Points on Thyroid Cysts and Tumors
✅ Thyroid cysts are often benign; tumors may be benign or malignant
✅ FNA biopsy is the gold standard for evaluation of thyroid nodules
✅ Papillary carcinoma is the most common and has excellent prognosis
✅ Early diagnosis and treatment reduce risk of complications
✅ Total thyroidectomy + RAI + levothyroxine is standard in many thyroid cancers
✅ Nurses play a key role in airway management, voice monitoring, calcium assessment, and patient education
🧠 Disorders of the Parathyroid Gland
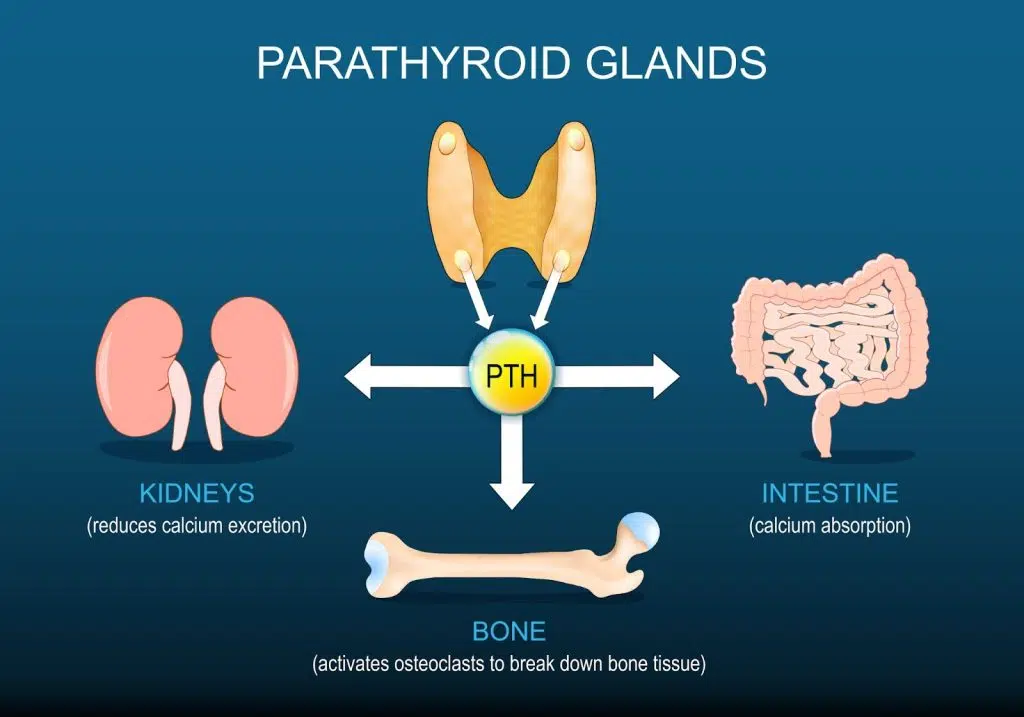
📌 Overview of the Parathyroid Gland:
- The parathyroid glands are four small glands located behind the thyroid gland.
- They secrete parathyroid hormone (PTH), which is vital for regulating calcium and phosphate balance in the blood.
- PTH increases blood calcium levels by:
- Stimulating bone resorption
- Enhancing calcium reabsorption in kidneys
- Promoting activation of Vitamin D, which increases intestinal absorption of calcium
🔍 Major Disorders of the Parathyroid Gland:
| 🔬 Disorder | ⬆️/⬇️ PTH | 📌 Description |
|---|---|---|
| Hyperparathyroidism | ↑ PTH | Excessive secretion of PTH leading to hypercalcemia |
| Hypoparathyroidism | ↓ PTH | Inadequate secretion of PTH causing hypocalcemia |
| Pseudohypoparathyroidism | Normal or ↑ PTH | Genetic condition where tissues are resistant to PTH |
⚠️ 1. Hyperparathyroidism
📌 Definition:
A condition where one or more parathyroid glands secrete excess PTH, leading to elevated blood calcium levels (hypercalcemia) and bone demineralization.
🎯 Causes:
| Type | Cause |
|---|---|
| Primary | Parathyroid adenoma (most common), hyperplasia, or carcinoma |
| Secondary | Chronic kidney disease (causes hypocalcemia → increased PTH) |
| Tertiary | Prolonged secondary hyperparathyroidism causing autonomous PTH secretion |
🧬 Pathophysiology:
Excess PTH →
⬆️ Bone resorption →
⬆️ Calcium released into blood →
⬇️ Bone density →
- ⬆️ Renal calcium reabsorption & ⬆️ intestinal absorption via vitamin D activation →
Hypercalcemia
🚨 Signs and Symptoms:
💡 Use the mnemonic: “Bones, Stones, Groans, and Moans”
- Bones: Bone pain, fractures, osteoporosis
- Stones: Kidney stones (nephrolithiasis)
- Groans: GI symptoms – nausea, constipation, abdominal pain
- Moans: CNS – fatigue, confusion, depression
- Polyuria, muscle weakness, hypertension
🧪 Diagnosis:
- ↑ PTH
- ↑ Serum calcium
- ↓ Serum phosphate
- Bone density scan (DEXA): shows osteoporosis
- Imaging: parathyroid scan (sestamibi), ultrasound, CT/MRI
💊 Medical Management:
- Hydration (IV fluids) to dilute calcium
- Bisphosphonates to reduce bone loss
- Calcimimetics (e.g., cinacalcet) to reduce PTH secretion
- Phosphate supplements (in secondary type)
- Monitor calcium and renal function regularly
✂️ Surgical Management:
- Parathyroidectomy – Removal of overactive gland(s)
- Autotransplantation – In cases of hyperplasia, part of gland transplanted to forearm
🩺 Nursing Management:
- Monitor serum calcium, phosphate, and PTH
- Encourage fluid intake
- Watch for signs of hypercalcemia
- Post-op: watch for hypocalcemia (tingling, Trousseau’s & Chvostek’s signs)
- Educate about dietary calcium/phosphorus, medication, and follow-ups
❄️ 2. Hypoparathyroidism
📌 Definition:
A condition caused by insufficient PTH secretion, leading to hypocalcemia and hyperphosphatemia.
🎯 Causes:
| Type | Cause |
|---|---|
| Acquired | Accidental removal/damage during thyroid/parathyroid surgery (most common) |
| Autoimmune | Isolated or part of autoimmune polyendocrine syndromes |
| Congenital | DiGeorge syndrome (absent glands) |
| Radiation-induced | Neck or thyroid radiation therapy |
🧬 Pathophysiology:
↓ PTH →
↓ Calcium reabsorption from bone, kidney, intestine →
⬇️ Serum calcium →
⬆️ Serum phosphate →
Neuromuscular irritability and tetany
🚨 Signs and Symptoms:
| 🧠 System | 🔍 Symptoms |
|---|---|
| Neuromuscular | Tetany, tingling (fingers, lips), cramps, spasms |
| Positive signs | Trousseau’s sign (carpal spasm), Chvostek’s sign (facial twitching) |
| CNS | Anxiety, irritability, seizures |
| Cardiac | Arrhythmias, hypotension |
| Other | Brittle nails, dry skin, hair loss, dental hypoplasia |
🧪 Diagnosis:
- ↓ PTH
- ↓ Serum calcium
- ↑ Serum phosphate
- ECG: prolonged QT interval
- Check vitamin D and magnesium levels
💊 Medical Management:
- Calcium supplements (oral or IV in acute cases)
- Vitamin D analogs (calcitriol)
- Magnesium supplementation if low
- High-calcium, low-phosphate diet
- Thiazide diuretics (reduce calcium loss in urine)
🩺 Nursing Management:
- Monitor calcium, phosphate, PTH levels regularly
- Observe for signs of hypocalcemia (muscle twitching, tetany)
- Ensure seizure precautions if calcium is critically low
- Provide calcium-rich diet and limit high-phosphate foods
- Educate on lifelong therapy, medication adherence, and emergency signs
⚠️ 3. Pseudohypoparathyroidism
📌 Definition:
A rare genetic disorder in which body tissues are resistant to PTH, despite normal or elevated hormone levels.
🧬 Pathophysiology:
- PTH is produced normally, but target tissues don’t respond, causing hypocalcemia and hyperphosphatemia.
Symptoms:
- Similar to hypoparathyroidism: tetany, muscle cramps
- Short stature, round face, developmental delays
- Known as Albright hereditary osteodystrophy
Diagnosis:
- ↑ PTH
- ↓ Calcium
- ↑ Phosphate
- Genetic testing
Management:
- Calcium and vitamin D supplementation
- Symptom management and genetic counseling
🧷 Key Points on Parathyroid Disorders
✅ Parathyroid glands regulate serum calcium and phosphate via PTH
✅ Hyperparathyroidism causes hypercalcemia, kidney stones, bone loss
✅ Hypoparathyroidism causes hypocalcemia, tetany, muscle spasms
✅ Surgery (thyroidectomy) is the most common cause of acquired hypoparathyroidism
✅ Famous signs in hypocalcemia: Trousseau’s and Chvostek’s signs
✅ Medical management includes calcium, vitamin D, bisphosphonates, calcimimetics
✅ Nursing role involves calcium monitoring, seizure precautions, and patient education
❄️ HYPOPARATHYROIDISM
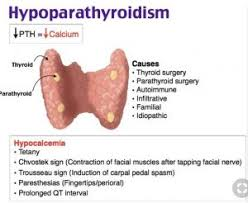
📌 Definition:
Hypoparathyroidism is a rare endocrine disorder characterized by inadequate secretion or action of parathyroid hormone (PTH), resulting in hypocalcemia (low blood calcium) and hyperphosphatemia (high blood phosphate).
🧠 Since PTH plays a critical role in maintaining calcium homeostasis, its deficiency leads to neuromuscular excitability, muscle cramps, tetany, and seizures.
⚠️ Causes of Hypoparathyroidism:
| 🎯 Cause | 📋 Description |
|---|---|
| Surgical (most common) | Accidental removal or damage of parathyroid glands during thyroidectomy, parathyroidectomy, or neck surgery |
| Autoimmune | Autoimmune destruction of parathyroid tissue, often part of Autoimmune Polyendocrine Syndrome (APS) |
| Congenital/Genetic | Developmental absence or hypoplasia of parathyroid glands (e.g., DiGeorge syndrome) |
| Radiation-induced | Radiation therapy to neck region damaging parathyroid glands |
| Infiltrative diseases | Hemochromatosis, Wilson’s disease, granulomas involving the parathyroids |
| Magnesium deficiency or excess | Affects PTH secretion and function |
🧬 Types of Hypoparathyroidism:
| 🔖 Type | 📖 Description |
|---|---|
| Acquired Hypoparathyroidism | Most common; occurs after surgery or radiation |
| Autoimmune Hypoparathyroidism | Due to autoantibodies against parathyroid glands; may be isolated or part of APS-1 |
| Congenital Hypoparathyroidism | Present at birth; e.g., DiGeorge syndrome (22q11 deletion) |
| Idiopathic Hypoparathyroidism | Cause unknown; diagnosis of exclusion |
| Pseudohypoparathyroidism | Rare genetic disorder where PTH is present but target tissues are resistant to it (not true hormone deficiency) |
🧬 Pathophysiology of Hypoparathyroidism:
- 🔻 Decreased PTH Secretion or Action:
- In hypoparathyroidism, parathyroid hormone (PTH) is either absent, decreased, or ineffective (e.g., in pseudohypoparathyroidism).
- ❌ Disrupted Calcium Regulation:
- PTH normally maintains calcium levels by:
- Stimulating bone resorption
- Increasing renal calcium reabsorption
- Promoting activation of vitamin D → increases intestinal calcium absorption
- Without PTH, all these processes decrease, leading to hypocalcemia.
- PTH normally maintains calcium levels by:
- 📈 Phosphate Retention:
- PTH also promotes phosphate excretion.
- Low PTH → decreased phosphate excretion → hyperphosphatemia
- ⚡ Neuromuscular Excitability:
- Hypocalcemia increases neuromuscular excitability, leading to:
- Tetany
- Muscle cramps
- Seizures
- Cardiac arrhythmias
- Hypocalcemia increases neuromuscular excitability, leading to:
🚨 Signs and Symptoms of Hypoparathyroidism:
🧠 Symptoms result primarily from low serum calcium and high phosphate levels.
🦴 Neuromuscular Signs (Classic Features):
| 🔍 Sign | 📝 Description |
|---|---|
| Tetany | Involuntary muscle spasms, cramps (especially in hands and feet) |
| Trousseau’s Sign | Carpal spasm induced by inflating a BP cuff |
| Chvostek’s Sign | Facial twitching when tapping the facial nerve |
| Paresthesia | Numbness and tingling, especially around the mouth, fingers, and toes |
| Muscle stiffness | Painful cramps and rigidity in extremities |
🧠 CNS Symptoms:
- Anxiety, irritability
- Confusion or memory impairment
- Seizures (in severe cases)
💓 Cardiac Symptoms:
- Prolonged QT interval on ECG
- Arrhythmias
- Hypotension
- Decreased myocardial contractility
🧍♀️ Other Symptoms:
- Dry skin, brittle nails
- Hair loss (especially on the scalp, eyebrows)
- Cataracts
- Dental abnormalities (in children)
- Laryngeal or bronchial spasms (in severe cases)
🧪 Diagnosis of Hypoparathyroidism:
| 🔬 Test | 📋 Interpretation |
|---|---|
| ✅ Serum Calcium | ↓ Decreased (hypocalcemia) |
| ✅ Serum Phosphate | ↑ Elevated (hyperphosphatemia) |
| ✅ Serum PTH | ↓ Decreased or absent (primary hypoparathyroidism) |
| ✅ Serum Magnesium | May be low; essential for PTH release |
| ✅ Vitamin D levels | May be low (active form, calcitriol) |
| ✅ ECG | Prolonged QT interval, arrhythmias |
| ✅ Urinary Calcium | May be elevated with supplementation or renal issues |
🔎 In Pseudohypoparathyroidism:
PTH levels are normal or high, but calcium remains low due to tissue resistance.
💊 I. MEDICAL MANAGEMENT
🎯 Goals of treatment:
- Restore and maintain normal serum calcium levels
- Correct hypocalcemia and hyperphosphatemia
- Prevent complications like tetany, seizures, and cardiac issues
- Provide lifelong hormone and mineral support (if chronic)
✅ 1. Acute Management (Emergency Hypocalcemia):
🚨 Used when patients present with severe hypocalcemia, tetany, seizures, or arrhythmias
| 🏥 Treatment | 📋 Description |
|---|---|
| IV calcium gluconate (10%) | Slowly infused under ECG monitoring; used for acute tetany or seizures |
| Magnesium sulfate IV (if deficient) | Essential for PTH secretion and calcium regulation |
| Airway monitoring and seizure precautions | In case of laryngeal spasms or convulsions |
✅ 2. Chronic/Long-term Management:
For maintenance of normal calcium-phosphorus balance and prevention of complications
| 💊 Medication/Therapy | 💡 Purpose |
|---|---|
| Oral calcium supplements (calcium carbonate/citrate) | Maintain serum calcium |
| Active Vitamin D (Calcitriol or Alfacalcidol) | Promotes calcium absorption in the intestine |
| Thiazide diuretics (e.g., hydrochlorothiazide) | Decreases urinary calcium loss |
| Low-phosphate diet | Reduces hyperphosphatemia |
| Phosphate binders (e.g., sevelamer) | Occasionally used if phosphate is persistently high |
| Recombinant PTH therapy (e.g., Natpara) | Used in selected cases of chronic hypoparathyroidism not controlled by supplements |
🔎 Monitoring During Treatment:
- Regular serum calcium, phosphate, magnesium, and PTH levels
- Urinary calcium excretion to avoid hypercalciuria and kidney stones
- ECG for arrhythmias if hypocalcemia is severe
🛠️ II. SURGICAL MANAGEMENT
❗ Hypoparathyroidism is not primarily treated surgically, but surgery may be involved in some specific situations:
🔪 Surgical Situations Involving Hypoparathyroidism:
| ⚠️ Surgical Context | 📋 Details |
|---|---|
| Parathyroid autotransplantation | During thyroidectomy or parathyroidectomy, healthy tissue is implanted into the forearm or neck to preserve function |
| Parathyroid gland reimplantation | In cases where removed glands were preserved |
| Thymus/parathyroid exploration | For congenital absence (DiGeorge syndrome) or genetic anomalies |
| Surgical correction of complications | E.g., removal of calcifications, cataracts due to chronic hypocalcemia |
🩺 Nursing Role in Surgical Cases:
- Monitor for hypocalcemia post-thyroid/parathyroid surgery
- Observe for Trousseau’s and Chvostek’s signs
- Ensure IV calcium is ready for emergency use
- Educate patient about lifelong follow-up, calcium/Vitamin D intake
🩺 NURSING MANAGEMENT OF HYPOPARATHYROIDISM
🎯 Goals of Nursing Care:
- Prevent and manage acute hypocalcemic episodes
- Monitor and correct electrolyte imbalances
- Promote patient safety and reduce neuromuscular complications
- Educate patient and family about lifelong treatment and self-care
🗂️ I. Nursing Assessment
| 🔍 Area | ✅ Key Assessment Points |
|---|---|
| Neuromuscular status | Monitor for tetany, muscle cramps, twitching, numbness/tingling |
| Trousseau’s sign | Inflate BP cuff → carpal spasm = positive |
| Chvostek’s sign | Tap facial nerve → facial twitching = positive |
| Vital signs | Watch for bradycardia, hypotension, respiratory distress |
| Calcium and phosphate levels | Regularly monitor lab values |
| ECG | Look for prolonged QT interval or arrhythmias |
| Seizure activity | Watch for changes in LOC or convulsions in severe hypocalcemia |
📝 II. Common Nursing Diagnoses
- Risk for electrolyte imbalance related to decreased PTH secretion
- Risk for injury (seizures) related to hypocalcemia
- Ineffective breathing pattern related to laryngeal spasm (in acute hypocalcemia)
- Acute pain related to muscle cramps and tetany
- Deficient knowledge regarding disease management, medication, and dietary needs
🧾 III. Nursing Interventions
🔹 For Acute Hypocalcemia (Emergency Care):
| 💡 Intervention | 🩺 Rationale |
|---|---|
| Administer IV calcium gluconate as prescribed | Corrects severe hypocalcemia quickly |
| Monitor ECG continuously | Detect arrhythmias due to electrolyte imbalance |
| Maintain seizure precautions | Prevent injury from convulsions |
| Provide calm, quiet environment | Reduces muscle excitability and stress-induced spasms |
| Ensure airway equipment is available | In case of laryngeal or bronchial spasm |
🔹 For Long-Term/Chronic Care:
| 💡 Intervention | 🩺 Rationale |
|---|---|
| Administer oral calcium and active Vitamin D supplements | Maintains calcium homeostasis |
| Educate on symptoms of hypocalcemia and hypercalcemia | Promotes early detection and self-monitoring |
| Monitor calcium and phosphate levels regularly | Ensures appropriate dosing and prevents complications |
| Encourage compliance with follow-up visits | Needed to adjust medication and monitor bone/kidney health |
| Teach about diet rich in calcium and low in phosphate | Supports overall management (e.g., avoid high-phosphate foods like dairy, meat, soft drinks) |
🧑🏫 IV. Patient and Family Education
- 💊 Lifelong need for calcium and vitamin D supplements
- 📈 Importance of regular blood tests (calcium, phosphate, PTH)
- 🧀 Dietary tips: Include calcium-rich, low-phosphorus foods
- ⚠️ Teach warning signs of:
- Hypocalcemia: tingling, cramps, spasms
- Hypercalcemia (due to over-supplementation): weakness, nausea, confusion
- 📅 Keep consistent follow-up appointments with endocrinologist
- 💡 Use a medication diary to track dosages and symptoms
📊 V. Evaluation Criteria (Expected Outcomes)
- Patient remains free from tetany or seizures
- Serum calcium and phosphate levels are within normal range
- Patient demonstrates correct medication use and reports compliance
- Patient and family express understanding of disease and management
- No evidence of cardiac or respiratory complications
⚠️ COMPLICATIONS OF HYPOPARATHYROIDISM
If not treated or monitored properly, hypoparathyroidism can lead to serious short-term and long-term complications:
🔥 1. Acute Hypocalcemic Crisis
- Sudden and severe drop in calcium levels
- Symptoms: laryngeal spasm, bronchospasm, seizures, cardiac arrhythmias
- May lead to respiratory arrest or death if not promptly treated
💓 2. Cardiac Complications
- Prolonged QT interval on ECG
- Risk of arrhythmias (e.g., torsades de pointes)
- Decreased myocardial contractility → heart failure (in chronic cases)
🧠 3. Neuromuscular Complications
- Chronic tetany and muscle spasms
- Seizures due to neuromuscular excitability
- Laryngeal spasm causing airway obstruction
👁️ 4. Ectopic Calcifications
- Due to long-standing hyperphosphatemia and calcium-phosphate imbalance
- Can affect:
- Brain (basal ganglia calcification → movement disorders)
- Eyes (cataracts)
- Kidneys (nephrocalcinosis or stones)
🦷 5. Dental Abnormalities (in children)
- Delayed tooth eruption
- Malformation of teeth
- Enamel hypoplasia
🧬 6. Psychosocial Effects
- Chronic fatigue, anxiety, depression
- Impact on quality of life due to lifelong medication dependence
🧷 KEY POINTS ON HYPOPARATHYROIDISM
✅ Definition: Deficiency or absence of parathyroid hormone (PTH) → hypocalcemia + hyperphosphatemia
✅ Common Causes:
- Surgical removal of parathyroid glands (most common)
- Autoimmune, congenital, or radiation-induced damage
✅ Types:
- Acquired, autoimmune, congenital, pseudohypoparathyroidism
✅ Classic Signs:
- Trousseau’s sign, Chvostek’s sign, tetany, paresthesia, seizures
✅ Lab Findings:
- ↓ Calcium, ↓ PTH, ↑ Phosphate
- ECG: prolonged QT interval
✅ Management:
- IV calcium gluconate in emergency
- Oral calcium + active vitamin D (calcitriol) long-term
- Thiazide diuretics, phosphate binders, magnesium if needed
✅ Nursing Focus:
- Monitor calcium levels
- Prevent tetany and seizures
- Patient education on lifelong therapy, diet, and self-monitoring
✅ Complications to Watch:
- Hypocalcemic crisis, cardiac arrhythmias, seizures, calcifications, cataracts
🔥 HYPERPARATHYROIDISM
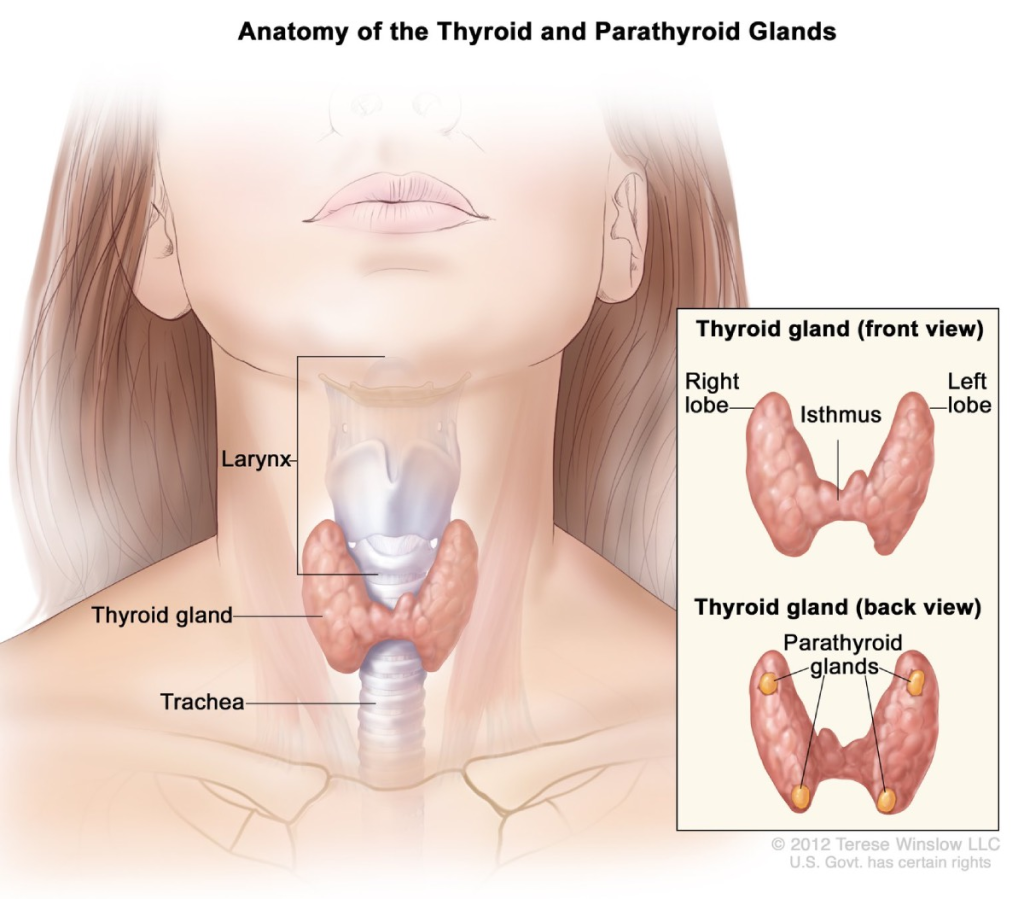
📌 Definition:
Hyperparathyroidism is a condition characterized by excessive secretion of parathyroid hormone (PTH) by one or more of the parathyroid glands, leading to elevated blood calcium levels (hypercalcemia) and low phosphate levels (hypophosphatemia).
🧠 PTH regulates calcium and phosphate. Excess PTH results in increased bone resorption, renal calcium reabsorption, and intestinal calcium absorption, causing hypercalcemia.
🎯 Causes of Hyperparathyroidism:
| 🩺 Cause | 📋 Description |
|---|---|
| Parathyroid adenoma | Most common cause (85% of cases); a benign tumor on one gland |
| Parathyroid hyperplasia | All four glands are enlarged and overactive |
| Parathyroid carcinoma | Rare cause of very high calcium and PTH levels |
| Chronic kidney disease | Causes secondary hyperparathyroidism due to low calcium and high phosphate |
| Vitamin D deficiency | Leads to compensatory PTH increase |
| Malabsorption syndromes | Reduced calcium absorption triggers PTH release |
| Genetic mutations | Seen in familial hyperparathyroid syndromes (MEN 1, MEN 2A) |
🧬 Types of Hyperparathyroidism:
| 🔖 Type | 📖 Description |
|---|---|
| Primary Hyperparathyroidism | Overproduction of PTH due to gland abnormality (e.g., adenoma, hyperplasia, cancer) |
| Secondary Hyperparathyroidism | Occurs as a response to chronic hypocalcemia (usually due to chronic kidney disease or vitamin D deficiency); glands are normal but overactive |
| Tertiary Hyperparathyroidism | Long-standing secondary hyperparathyroidism becomes autonomous and continues even after calcium/phosphate correction (common in ESRD patients) |
🧬 Pathophysiology:
- Overproduction of PTH:
- One or more parathyroid glands secrete excessive parathyroid hormone (PTH), often due to adenoma, hyperplasia, or chronic hypocalcemia.
- Bone Effects (↑ Resorption):
- PTH stimulates osteoclast activity, breaking down bone to release calcium → bone demineralization and osteoporosis
- Kidney Effects (↑ Reabsorption of Calcium):
- Increased calcium reabsorption → hypercalcemia
- Increased phosphate excretion → hypophosphatemia
- Also increases risk of renal calculi (kidney stones)
- Intestinal Effects (↑ Absorption):
- PTH stimulates activation of vitamin D → increases calcium absorption from the gut
- Chronic Result:
- Sustained hypercalcemia, bone weakness, kidney damage, and possible neurological and GI disturbances
🚨 Signs and Symptoms:
Mnemonic: “Bones, Stones, Groans, and Moans”
🦴 Bones – Musculoskeletal Symptoms:
- Bone pain, tenderness
- Fractures due to osteoporosis
- Muscle weakness
💎 Stones – Renal Symptoms:
- Kidney stones (nephrolithiasis)
- Polyuria and polydipsia
- Dehydration
😣 Groans – Gastrointestinal Symptoms:
- Nausea, vomiting
- Constipation
- Abdominal pain
- Pancreatitis (in severe cases)
😫 Moans – Neurological/Psychiatric Symptoms:
- Fatigue, depression
- Confusion or memory issues
- Irritability
- Sleep disturbances
- In severe cases: stupor or coma
💓 Cardiovascular Symptoms:
- Hypertension
- Arrhythmias (due to hypercalcemia)
🧪 Diagnosis:
| 🔬 Test | 📋 Expected Results |
|---|---|
| ✅ Serum calcium | ↑ Elevated (>10.5 mg/dL) |
| ✅ Serum phosphate | ↓ Decreased |
| ✅ Serum PTH | ↑ Elevated (in primary/tertiary) |
| ✅ Vitamin D levels | May be low in secondary hyperparathyroidism |
| ✅ 24-hour urine calcium | ↑ Elevated (increased calcium excretion) |
| ✅ Bone density scan (DEXA) | ↓ Bone mineral density, osteoporosis |
| ✅ Renal ultrasound or CT | Detects kidney stones or nephrocalcinosis |
| ✅ Sestamibi scan/Parathyroid scan | Localizes adenoma or hyperplasia |
| ✅ ECG | May show shortened QT interval (due to hypercalcemia) |
💊 I. MEDICAL MANAGEMENT
🎯 Goals of Medical Management:
- Normalize serum calcium and phosphate levels
- Preserve bone density
- Prevent kidney stones and renal damage
- Prepare for or avoid surgery when possible
🔹 1. Hydration Therapy
| 💧 Treatment | 💡 Purpose |
|---|---|
| Oral/IV fluids (Normal Saline) | Dilutes serum calcium and promotes renal excretion |
🔹 2. Medications
| 💊 Drug Class | Examples | 📋 Purpose |
|---|---|---|
| Bisphosphonates | Alendronate, Pamidronate | Inhibit bone resorption; improve bone density |
| Calcimimetics | Cinacalcet | Reduce PTH secretion by mimicking calcium; used in secondary/tertiary types |
| Loop diuretics | Furosemide | Increases calcium excretion (never use thiazides—they increase calcium) |
| Vitamin D analogs | Calcitriol | Used in secondary hyperparathyroidism to suppress PTH |
| Phosphate binders | Sevelamer, calcium acetate | Reduce serum phosphate levels in CKD patients |
| Estrogen or Raloxifene | Postmenopausal women | Help maintain bone mineral density |
🔎 Monitoring:
- Serum calcium, phosphate, PTH
- Renal function
- Bone density (DEXA)
- Urine calcium to avoid hypercalciuria and stones
🛠️ II. SURGICAL MANAGEMENT
🎯 Parathyroidectomy (removal of overactive parathyroid gland/s) is the definitive treatment for primary and tertiary hyperparathyroidism.
✂️ Types of Surgical Options:
| Surgery | 📋 Description |
|---|---|
| Focused (minimally invasive) parathyroidectomy | Removal of identified adenoma (most common type) |
| Subtotal parathyroidectomy | Removal of 3½ glands in cases of hyperplasia |
| Total parathyroidectomy with autotransplantation | All glands removed; part of one gland transplanted into forearm or neck muscle (to preserve some function) |
🔍 Indications for Surgery:
- Symptomatic hypercalcemia (bones, stones, groans)
- Serum calcium >1 mg/dL above normal
- Reduced kidney function (eGFR <60 mL/min)
- Osteoporosis on DEXA scan
- Age <50 years (due to long-term risk)
- Presence of kidney stones or calcifications
- Parathyroid carcinoma (rare but aggressive)
🩺 Preoperative Considerations:
- Hydrate patient adequately to reduce calcium
- Correct electrolyte imbalances
- Perform localization studies (ultrasound, sestamibi scan)
🛏️ Postoperative Care:
| Focus Area | Nursing Responsibility |
|---|---|
| Monitor for hypocalcemia | Common after gland removal → watch for Trousseau’s and Chvostek’s signs |
| Serum calcium and PTH levels | Check frequently for early drop |
| IV calcium gluconate | Keep ready for emergency hypocalcemia |
| Airway monitoring | Especially after neck surgery; watch for swelling or stridor |
| Voice assessment | Recurrent laryngeal nerve injury can cause hoarseness |
🩺 NURSING MANAGEMENT OF HYPERPARATHYROIDISM
🎯 Nursing Goals:
- Normalize serum calcium levels
- Monitor and prevent complications (e.g., bone loss, kidney stones)
- Prepare and support the patient through medical and surgical treatment
- Promote medication adherence and lifestyle changes
- Educate about long-term follow-up and self-care
🗂️ I. Nursing Assessment
| 🔍 Area | ✅ Key Assessment Points |
|---|---|
| Neurological status | Monitor for confusion, lethargy, muscle weakness |
| GI symptoms | Constipation, abdominal pain, nausea |
| Skeletal symptoms | Bone pain, fractures, reduced height |
| Renal function | Monitor for signs of kidney stones: flank pain, hematuria |
| Hydration status | Assess for signs of dehydration (dry mucosa, hypotension) |
| Lab monitoring | Serum calcium, phosphate, PTH, renal function, and vitamin D |
| ECG | Watch for shortened QT interval, arrhythmias |
📝 II. Common Nursing Diagnoses
- Risk for electrolyte imbalance (related to elevated calcium)
- Impaired urinary elimination (related to renal stones)
- Risk for injury (related to bone demineralization and weakness)
- Impaired physical mobility (due to bone pain/fractures)
- Knowledge deficit (related to disease process and treatment)
🧾 III. Nursing Interventions
🔹 For Medical Management:
| 💡 Intervention | 🩺 Rationale |
|---|---|
| Encourage high fluid intake (2.5–3 L/day) | Prevent kidney stones and promote calcium excretion |
| Administer loop diuretics (e.g., furosemide) as ordered | Promote calcium excretion (never give thiazides) |
| Monitor serum calcium, phosphate, and PTH levels regularly | Evaluate effectiveness of therapy and detect complications early |
| Administer bisphosphonates or calcimimetics as prescribed | To reduce bone resorption and lower calcium levels |
| Promote weight-bearing exercises (if tolerated) | Helps strengthen bones and reduce bone loss |
| Encourage low-calcium, high-fiber diet (unless otherwise directed) | Manages constipation and calcium intake if needed |
🔹 For Surgical Patients (Post-Parathyroidectomy):
| 🩺 Post-op Focus | Nursing Action |
|---|---|
| Monitor for hypocalcemia | Look for Trousseau’s and Chvostek’s signs, tingling around mouth/fingers |
| Airway observation | Watch for signs of swelling, stridor, or respiratory distress |
| Voice changes | May indicate recurrent laryngeal nerve injury |
| Pain management | Provide analgesics and reassure patient |
| IV calcium gluconate at bedside | Keep ready for emergency hypocalcemia |
| Patient positioning | Keep in semi-Fowler’s to reduce swelling and aid breathing |
🧑🏫 IV. Patient and Family Education
- 🚰 Hydration: Drink plenty of fluids to prevent stones
- 🥦 Diet: Follow dietary advice (may vary by type and treatment phase)
- 💊 Medication adherence: Take prescribed calcium-lowering meds or supplements regularly
- 🧪 Lab monitoring: Regular testing of calcium, PTH, vitamin D, and renal function is essential
- 🧠 Signs to report: Confusion, muscle twitching, bone pain, or signs of kidney stones
- 🗓️ Follow-up: Lifelong monitoring may be required, especially after surgery
📊 V. Evaluation Criteria (Expected Outcomes)
- Serum calcium and phosphate levels maintained within normal range
- Patient remains free from fractures, kidney stones, and neurological symptoms
- Patient demonstrates knowledge of disease and treatment plan
- Patient reports improved comfort, energy, and mobility
- No signs of hypocalcemia or hypercalcemia crisis post-surgery
⚠️ COMPLICATIONS OF HYPERPARATHYROIDISM
If left untreated or poorly managed, hyperparathyroidism can lead to multi-system complications due to persistently high calcium levels and bone demineralization.
🦴 1. Skeletal Complications:
- Osteoporosis and osteopenia
- Bone pain, fragility fractures, especially in the spine and long bones
- Osteitis fibrosa cystica (rare): bone lesions, cysts due to extreme bone resorption
💎 2. Renal Complications:
- Nephrolithiasis (kidney stones) from hypercalciuria
- Nephrocalcinosis (calcium deposits in kidneys)
- Progressive renal insufficiency or chronic kidney disease (CKD)
💓 3. Cardiovascular Complications:
- Hypertension
- Arrhythmias due to altered calcium-potassium balance
- Shortened QT interval on ECG
- Vascular and valvular calcification (especially in secondary hyperparathyroidism)
😫 4. Neurological and Psychiatric Complications:
- Cognitive impairment, memory loss
- Depression, anxiety
- Fatigue, lethargy
- Seizures (rare, usually from very high calcium levels)
🍽️ 5. Gastrointestinal Complications:
- Peptic ulcers, constipation, nausea, and pancreatitis
🧪 6. Postoperative Complication: Hypocalcemia
- “Hungry bone syndrome”: sudden drop in calcium post-parathyroidectomy due to rapid bone remineralization
- Requires urgent calcium supplementation
🧷 KEY POINTS ON HYPERPARATHYROIDISM
✅ Definition: Excess production of PTH by the parathyroid glands, causing hypercalcemia and hypophosphatemia
✅ Common Causes:
- Primary: Parathyroid adenoma (most common)
- Secondary: Chronic kidney disease, vitamin D deficiency
- Tertiary: Autonomous PTH secretion after prolonged secondary hyperparathyroidism
✅ Classic Symptoms Mnemonic:
“Bones, Stones, Groans, and Moans”
→ Bone pain, kidney stones, GI upset, and neuropsychiatric symptoms
✅ Diagnosis:
- ↑ Serum calcium, ↑ PTH, ↓ phosphate
- Bone density scan (DEXA), sestamibi scan, renal ultrasound
✅ Medical Treatment:
- Hydration, loop diuretics, bisphosphonates, calcimimetics, vitamin D
✅ Surgical Treatment:
- Parathyroidectomy (minimally invasive, subtotal, or total with autotransplantation)
✅ Nursing Role:
- Monitor calcium levels
- Watch for signs of hyper- and hypocalcemia
- Educate on diet, hydration, medications, and follow-up
✅ Complications:
- Bone loss, kidney stones, cardiac arrhythmias, neuropsychiatric changes, and post-op hypocalcemia
🧠 MYXEDEMA
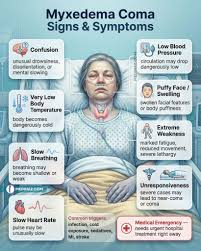
📌 Definition:
Myxedema is a severe and advanced form of hypothyroidism, characterized by the accumulation of mucopolysaccharides in the skin and other tissues, causing non-pitting edema, especially in the face, hands, and feet.
🧬 It represents the end-stage of untreated or poorly managed hypothyroidism.
In extreme cases, it may lead to myxedema coma—a life-threatening emergency.
🎯 Causes of Myxedema:
| 🔍 Cause | 📋 Description |
|---|---|
| Untreated or long-standing hypothyroidism | Most common cause |
| Autoimmune thyroiditis (Hashimoto’s disease) | Destruction of thyroid tissue |
| Post-thyroidectomy | Without adequate hormone replacement |
| Radiation therapy | To neck or thyroid |
| Drug-induced | Lithium, amiodarone, antithyroid medications |
| Pituitary or hypothalamic failure | Secondary or tertiary hypothyroidism |
| Severe illness/infection in hypothyroid patients | Triggers myxedema crisis |
🧾 Types of Myxedema:
| 🔖 Type | 📖 Description |
|---|---|
| Primary Myxedema | Due to direct thyroid gland failure (e.g., autoimmune destruction) |
| Secondary Myxedema | Due to pituitary failure → ↓ TSH secretion |
| Tertiary Myxedema | Due to hypothalamic dysfunction → ↓ TRH → ↓ TSH |
| Myxedema Coma | Life-threatening complication of severe untreated hypothyroidism, often precipitated by infection, surgery, or trauma |
🔬 Pathophysiology:
- 📉 Severe deficiency of thyroid hormones (T3, T4)
- 🔄 Slowing of metabolic rate in all body systems
- 📥 Accumulation of glycosaminoglycans (mucopolysaccharides) in interstitial tissues → non-pitting edema
- 🧠 Depression of neurological and cardiovascular function
- ❄️ Hypothermia, hypoventilation, bradycardia, hyponatremia → can progress to coma and death
🚨 Signs and Symptoms:
| ⚠️ System | 🔍 Symptoms |
|---|---|
| General | Fatigue, lethargy, cold intolerance, weight gain, puffy face |
| Skin | Dry, coarse skin, non-pitting edema (face, hands, feet), pale appearance |
| Hair & Nails | Hair thinning, loss of outer third of eyebrows, brittle nails |
| Cardiovascular | Bradycardia, hypotension, enlarged heart (cardiomegaly) |
| Respiratory | Hypoventilation, dyspnea |
| Neurological | Slow mental activity, confusion, memory loss, depression |
| Gastrointestinal | Constipation, anorexia |
| Reproductive | Menstrual irregularities, infertility |
| In Myxedema Coma | Hypothermia, stupor/coma, respiratory failure, hypoglycemia, seizures |
🧪 Diagnosis:
| 🔬 Test | 📋 Findings |
|---|---|
| Serum TSH | ↑ in primary myxedema, ↓ in secondary/tertiary |
| Free T3 and T4 | ↓ Decreased |
| Serum sodium | ↓ Hyponatremia (in severe cases) |
| Serum glucose | ↓ Hypoglycemia |
| ABG | Respiratory acidosis (in coma) |
| ECG | Bradycardia, low voltage QRS, prolonged QT |
| Brain imaging | Rule out other coma causes in severe cases |
💊 Medical Management:
| 💊 Treatment | 📋 Purpose |
|---|---|
| Levothyroxine (T4) | Lifelong hormone replacement; start low, go slow |
| Liothyronine (T3) | Given IV in myxedema coma for rapid action |
| Glucocorticoids | Used initially to prevent adrenal insufficiency (Hydrocortisone) |
| Supportive care | Oxygen, warming, glucose infusion, fluid/electrolyte correction |
| Treatment of underlying cause | Infection, trauma, drug withdrawal, etc. |
✂️ Surgical Management:
❌ Surgery is not a treatment for myxedema itself, but may be indicated in:
- Post-thyroidectomy myxedema due to inadequate hormone replacement
- Tumors of pituitary or hypothalamus causing secondary or tertiary hypothyroidism
- Thyroidectomy in Hashimoto’s with large goiter (very rare)
🩺 Nursing Management:
| 🎯 Focus Area | 🩺 Nursing Action |
|---|---|
| Vital signs monitoring | Observe for bradycardia, hypotension, hypothermia |
| Neurological status | Monitor LOC, confusion, drowsiness |
| Respiratory care | Watch for shallow breathing, provide oxygen if needed |
| Administer medications | Levothyroxine as prescribed; monitor effects |
| Prevent skin breakdown | Turn frequently, keep skin clean and moisturized |
| Nutritional support | Encourage warm fluids, fiber-rich diet |
| Patient education | Lifelong hormone therapy, signs of overdose (palpitations, sweating), need for follow-up |
| Emergency preparedness | Recognize signs of myxedema coma; keep IV access ready for emergencies |
⚠️ Complications:
- Myxedema coma (life-threatening)
- Hypothermia
- Hypoglycemia
- Cardiac arrhythmias or heart failure
- Respiratory failure
- Hyponatremia-induced seizures
- Permanent cognitive decline if untreated
🧷 Key Points Summary
✅ Myxedema is a severe, life-threatening stage of hypothyroidism
✅ Caused by untreated thyroid disease, surgery, radiation, drugs, or autoimmune destruction
✅ Classic features: Puffy face, cold intolerance, bradycardia, dry skin, lethargy
✅ Myxedema coma is a medical emergency → needs IV T3/T4, steroids, fluids, and intensive care
✅ Diagnosed by ↓ T3/T4, ↑ TSH (in primary) and clinical symptoms
✅ Managed by lifelong hormone replacement (levothyroxine)
✅ Nurses play a vital role in early detection, medication administration, monitoring, and education.
🧠 CRETINISM (CONGENITAL HYPOTHYROIDISM)

📌 Definition:
Cretinism is a condition of severe hypothyroidism present at birth or in early infancy, resulting in stunted physical growth and delayed mental development. It occurs due to deficiency or absence of thyroid hormones (T3, T4) during critical developmental periods.
If untreated, it leads to permanent intellectual disability, growth failure, and multiple system complications.
🧬 Causes of Cretinism:
| 🎯 Cause | 📋 Description |
|---|---|
| Congenital absence of thyroid gland (athyreosis) | No gland formed during fetal development |
| Ectopic thyroid gland | Thyroid tissue located outside normal position, poorly functioning |
| Thyroid dyshormonogenesis | Genetic defect in thyroid hormone production |
| Iodine deficiency (maternal or fetal) | Major cause in endemic areas |
| Maternal antithyroid drug use | Crosses placenta, suppresses fetal thyroid function |
| Maternal autoimmune disease | Antibodies cross placenta and attack fetal thyroid |
🏷️ Types of Cretinism:
| Type | Description |
|---|---|
| Neurological Cretinism | Intellectual disability, deaf-mutism, spasticity; due to fetal hypothyroidism |
| Myxedematous Cretinism | Growth retardation, coarse features, hypothyroid signs; often from postnatal iodine deficiency |
| Sporadic Cretinism | Due to genetic or developmental defects; occurs in iodine-sufficient areas |
| Endemic Cretinism | Occurs in iodine-deficient populations; preventable with iodization |
🔬 Pathophysiology:
- ↓ Thyroid hormones (T3, T4) during fetal/infant development
- Leads to impaired brain myelination and synaptic development
- Also causes delayed skeletal maturation and cartilage ossification
- Affects basal metabolism, protein synthesis, and growth hormones
- Results in mental retardation, short stature, skeletal deformities, and myxedematous features
🚨 Signs and Symptoms:
Often subtle at birth, become more apparent within first few months:
🧒 Infant Symptoms:
- Prolonged jaundice
- Constipation
- Poor feeding
- Hoarse cry
- Sleepiness, lethargy
- Hypotonia (floppy baby)
- Large fontanelles, wide sutures
- Macroglossia (large tongue)
- Puffy face, dry skin
- Umbilical hernia
- Poor growth and weight gain
👶 Later in Untreated Children:
- Stunted growth
- Delayed milestones
- Mental retardation
- Deaf-mutism
- Coarse facial features
- Dry skin, brittle hair
- Myxedematous face and extremities
🧪 Diagnosis:
| Test | Findings |
|---|---|
| Neonatal screening (TSH, T4) | Elevated TSH, low T4 (done within 48–72 hours after birth) |
| Serum thyroid profile | ↑ TSH (primary), ↓ T3, ↓ T4 |
| Thyroid scan (scintigraphy) | Shows absence, ectopic, or hypoplastic gland |
| Thyroid ultrasound | Detects gland size and position |
| X-ray (bone age) | Delayed epiphyseal maturation |
| Genetic testing | In suspected inherited enzyme defects |
💊 Medical Management:
Early treatment within the first 2 weeks of life is critical to prevent permanent damage.
| Treatment | Purpose |
|---|---|
| Levothyroxine (T4) | Start as early as possible, lifelong therapy |
| Initial dose | 10–15 mcg/kg/day (adjust based on labs) |
| Monitor | TSH, T4 every 2–4 weeks initially, then every 3–6 months |
| Supportive therapies | Speech therapy, physiotherapy, educational support for delays |
✂️ Surgical Management:
❌ Surgery is not indicated in cretinism. However, it may be used to treat:
- Goiter or compressive thyroid cysts (rare in congenital cases)
- Associated structural defects (e.g., umbilical hernia repair)
🩺 Nursing Management:
| Focus Area | Nursing Action |
|---|---|
| Early identification | Educate parents on neonatal screening importance |
| Administer levothyroxine | Crush tablets, mix with water or breast milk (not formula) |
| Monitor growth and development | Track milestones, weight, and height |
| Support feeding | Encourage frequent feeding in lethargic infants |
| Educate parents | Lifelong treatment, medication adherence, monitoring labs |
| Prevent complications | Watch for signs of undertreatment (lethargy, poor growth) |
| Refer for therapy | Speech, occupational, and developmental therapy as needed |
⚠️ Complications:
- Irreversible intellectual disability if untreated
- Growth retardation and short stature
- Deaf-mutism
- Delayed puberty
- Skeletal deformities
- Social and emotional difficulties
- Poor school performance
🧷 Key Points on Cretinism
✅ Cretinism = congenital hypothyroidism
✅ Caused by thyroid dysgenesis, iodine deficiency, or maternal antibodies/drugs
✅ Early signs: lethargy, constipation, macroglossia, poor growth
✅ Newborn screening is essential for early detection
✅ Start levothyroxine within 2 weeks of birth to prevent brain damage
✅ No surgical treatment needed; focus on lifelong hormone replacement
✅ Nurses must educate and support families for long-term management
🧬 Disorders of the Adrenal Gland
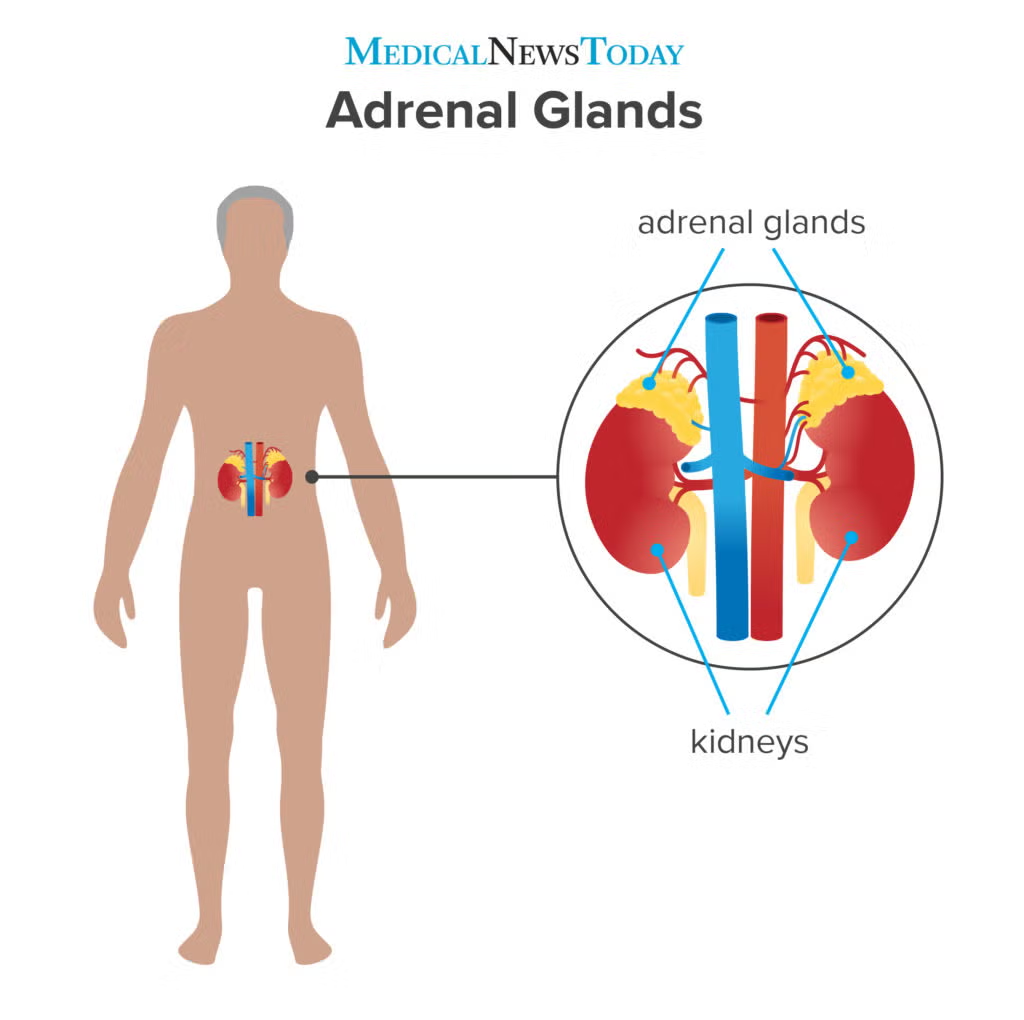
🏥 Overview of Adrenal Glands:
- The adrenal glands are small, triangular glands located on top of each kidney.
- Each gland has two parts:
- Adrenal cortex (outer layer): Produces cortisol, aldosterone, and androgens
- Adrenal medulla (inner layer): Produces catecholamines (adrenaline & noradrenaline)
Disorders may affect either or both layers and can lead to hypo- or hyperfunction.
🔍 Major Adrenal Disorders Include:
| 🔖 Disorder | 📋 Primary Hormone Affected |
|---|---|
| Addison’s Disease | ↓ Cortisol, ↓ Aldosterone |
| Cushing’s Syndrome | ↑ Cortisol |
| Congenital Adrenal Hyperplasia (CAH) | Abnormal androgens/cortisol |
| Pheochromocytoma | ↑ Adrenaline/Noradrenaline |
| Hyperaldosteronism (Conn’s Syndrome) | ↑ Aldosterone |
| Adrenal tumors | May affect any adrenal hormone |
| Adrenal insufficiency (acute or chronic) | Deficiency of adrenal hormones |
📌 1. Addison’s Disease (Primary Adrenal Insufficiency)
🧾 Definition:
Autoimmune or infectious destruction of adrenal cortex leading to deficiency of cortisol and aldosterone.
🧬 Causes:
- Autoimmune (most common)
- Tuberculosis, infections
- Surgical removal
- Adrenal hemorrhage
- Congenital adrenal hypoplasia
🔬 Symptoms:
- Weakness, fatigue
- Weight loss
- Hyperpigmentation
- Hypotension, dehydration
- Hyponatremia, hyperkalemia
- Salt craving
🧪 Diagnosis:
- ↓ Cortisol, ↑ ACTH
- ACTH stimulation test
- Electrolyte imbalance
💊 Management:
- Hydrocortisone (cortisol replacement)
- Fludrocortisone (aldosterone replacement)
- Emergency steroids during stress/infection
📌 2. Cushing’s Syndrome
🧾 Definition:
Excess cortisol in the body due to adrenal tumor, pituitary adenoma (Cushing’s disease), or steroid overuse.
🧬 Causes:
- Exogenous corticosteroids (most common)
- Pituitary tumor (Cushing’s disease)
- Adrenal adenoma/carcinoma
- Ectopic ACTH-producing tumor
🔬 Symptoms:
- Moon face, buffalo hump
- Truncal obesity, thin limbs
- Purple striae
- Hyperglycemia
- Hypertension
- Muscle wasting
- Osteoporosis
- Mood swings
🧪 Diagnosis:
- 24-hour urinary free cortisol
- Dexamethasone suppression test
- ACTH levels
- MRI/CT scan of pituitary/adrenal
💊 Management:
- Reduce or stop steroids (if iatrogenic)
- Surgery for tumors (pituitary or adrenal)
- Ketoconazole to inhibit cortisol synthesis
- Radiation (if tumor non-operable)
📌 3. Hyperaldosteronism (Conn’s Syndrome)
🧾 Definition:
Overproduction of aldosterone causing sodium retention and potassium loss.
🧬 Causes:
- Aldosterone-producing adenoma
- Bilateral adrenal hyperplasia
🔬 Symptoms:
- Hypertension
- Hypokalemia → fatigue, cramps, arrhythmias
- Polyuria, polydipsia
- Metabolic alkalosis
🧪 Diagnosis:
- ↑ Aldosterone, ↓ Renin
- Aldosterone-renin ratio
- CT/MRI adrenal imaging
💊 Management:
- Spironolactone or eplerenone (aldosterone antagonists)
- Adrenalectomy (if unilateral tumor)
📌 4. Pheochromocytoma
🧾 Definition:
A rare adrenal medulla tumor that secretes excess catecholamines (adrenaline & noradrenaline).
🧬 Causes:
- Usually benign tumor
- Associated with MEN 2A/2B, neurofibromatosis
🔬 Symptoms:
- Episodic hypertension
- Palpitations
- Headache, sweating
- Anxiety, tremors
- Hyperglycemia
🧪 Diagnosis:
- Plasma/urinary metanephrines and catecholamines
- CT/MRI scan
- MIBG scan (to locate tumors)
💊 Management:
- Alpha-blockers (e.g., phenoxybenzamine) pre-surgery
- Beta-blockers after alpha control
- Adrenalectomy
📌 5. Congenital Adrenal Hyperplasia (CAH)
🧾 Definition:
A group of inherited enzyme deficiencies causing abnormal cortisol synthesis and excess androgen production.
🧬 Causes:
- 21-hydroxylase deficiency (most common)
🔬 Symptoms:
- Ambiguous genitalia (females)
- Early puberty (males)
- Salt-wasting crisis (vomiting, dehydration)
- Short stature, acne
🧪 Diagnosis:
- ↑ 17-hydroxyprogesterone
- Electrolytes: ↓ sodium, ↑ potassium
- Genetic testing
💊 Management:
- Glucocorticoids (hydrocortisone)
- Mineralocorticoids (fludrocortisone)
- Salt supplements
- Surgery for genital anomalies (if needed)
🩺 General Nursing Management for Adrenal Disorders:
- Monitor vital signs, BP, blood sugar, and electrolytes
- Provide emotional support (due to mood changes, body image issues)
- Administer and monitor hormonal therapy
- Educate about lifelong medication adherence (especially Addison’s, CAH)
- Prepare for and assist during diagnostic tests (ACTH test, imaging)
- Provide high-calorie, low-sodium diet (Cushing’s), high-sodium diet (Addison’s)
⚠️ Complications (By Disorder):
| Condition | Complications |
|---|---|
| Addison’s | Addisonian crisis (shock, coma), arrhythmias |
| Cushing’s | Diabetes, osteoporosis, infections |
| Conn’s | Stroke, cardiac arrhythmia, renal damage |
| Pheochromocytoma | Hypertensive crisis, cardiac failure |
| CAH | Shock, infertility, electrolyte crisis |
🧷 Key Points Summary
✅ Adrenal gland = Cortex (cortisol, aldosterone, androgens) + Medulla (adrenaline)
✅ Hypofunction = Addison’s disease, adrenal crisis
✅ Hyperfunction = Cushing’s syndrome, Conn’s syndrome, pheochromocytoma
✅ Lifelong steroid therapy often required in chronic hypofunction
✅ Surgical removal indicated for tumors and hormone-producing adenomas
✅ Nurses must closely monitor for BP changes, electrolyte imbalances, and signs of hormone excess or deficiency
❄️ ADDISON’S DISEASE (Primary Adrenal Insufficiency)
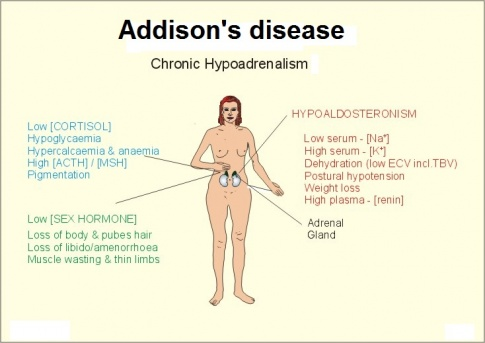
📌 Definition:
Addison’s disease is a rare endocrine disorder characterized by destruction or dysfunction of the adrenal cortex, leading to deficiency of cortisol, and often aldosterone and androgens as well.
🧠 It results in hypocortisolism, causing fatigue, weight loss, hypotension, and electrolyte imbalances.
🧬 Causes of Addison’s Disease:
| 🔍 Cause Category | 📋 Examples |
|---|---|
| Autoimmune destruction (most common) | Autoimmune adrenalitis – often part of Autoimmune Polyendocrine Syndrome (APS) |
| Infections | Tuberculosis (most common cause globally), fungal infections (e.g., histoplasmosis), CMV, HIV |
| Adrenal hemorrhage/infarction | Due to anticoagulants, trauma, or sepsis (Waterhouse-Friderichsen syndrome) |
| Surgical removal of adrenal glands | Bilateral adrenalectomy |
| Genetic or congenital causes | Congenital adrenal hypoplasia |
| Metastatic cancer to adrenal glands | Breast, lung, lymphoma |
| Drugs | Ketoconazole, rifampin (inhibit steroid synthesis) |
🧾 Types of Addison’s Disease:
| 🏷️ Type | 📖 Description |
|---|---|
| Primary Adrenal Insufficiency (True Addison’s Disease) | Destruction or dysfunction of the adrenal cortex itself → low cortisol + aldosterone |
| Secondary Adrenal Insufficiency | Due to pituitary failure → low ACTH → ↓ cortisol only (aldosterone usually normal) |
| Tertiary Adrenal Insufficiency | Due to hypothalamic dysfunction or sudden withdrawal of steroids (suppresses CRH and ACTH) |
| Acute Adrenal Insufficiency (Addisonian Crisis) | Sudden, life-threatening drop in cortisol and aldosterone levels due to stress, infection, or steroid withdrawal |
🧬 Pathophysiology of Addison’s Disease:
- Damage to the adrenal cortex (due to autoimmune disease, infection, or injury)
↓ - Decreased secretion of adrenal hormones:
- ↓ Cortisol → impaired glucose metabolism, stress response, and vascular tone
- ↓ Aldosterone → loss of sodium, water → hypotension; retention of potassium → hyperkalemia
- ↓ Androgens (in females) → reduced axillary/pubic hair, low libido
↓
- Increased ACTH (in primary type) due to lack of feedback inhibition
- High ACTH stimulates melanocytes, causing hyperpigmentation
↓
- High ACTH stimulates melanocytes, causing hyperpigmentation
- Overall effects:
- Low blood glucose (hypoglycemia)
- Low blood pressure (hypotension)
- Dehydration
- Electrolyte imbalance
- Fatigue and weakness
🚨 Signs and Symptoms of Addison’s Disease:
🟤 General Symptoms:
- Chronic fatigue, weakness
- Unexplained weight loss
- Anorexia, nausea, vomiting
- Low blood pressure (especially postural hypotension)
- Salt craving
- Irritability, depression
🟠 Skin Symptoms:
- Hyperpigmentation (bronzing of skin, especially on creases, elbows, knees, gums) – seen in primary Addison’s due to ↑ ACTH
🧪 Electrolyte-related Symptoms:
- Hyponatremia → dizziness, confusion
- Hyperkalemia → muscle weakness, cardiac arrhythmias
- Hypoglycemia → sweating, tremors, confusion
💓 Cardiovascular:
- Hypotension
- Weak pulse
- Risk of shock (especially during stress or illness)
🧑⚕️ Women-specific Symptoms:
- Decreased body hair (axillary and pubic)
- Reduced libido
⚠️ In Addisonian crisis (acute adrenal insufficiency), symptoms escalate rapidly:
- Severe hypotension or shock
- Confusion or coma
- Hypoglycemia
- Severe vomiting, diarrhea
- Emergency condition needing IV hydrocortisone + fluids
🧪 Diagnosis of Addison’s Disease
| 🧬 Test | 🔎 Interpretation |
|---|---|
| ✅ Serum cortisol | ↓ Low cortisol, especially in the early morning (normal: 6–8 a.m.) |
| ✅ Serum ACTH | ↑ High in primary Addison’s (due to lack of negative feedback) |
| ↓ or normal in secondary/tertiary adrenal insufficiency | |
| ✅ ACTH Stimulation Test (Cosyntropin Test) | Gold standard test: |
- No rise in cortisol after synthetic ACTH = primary adrenal insufficiency | | ✅ Electrolytes | ↓ Sodium, ↑ Potassium, ↓ Glucose, ↑ BUN | | ✅ Plasma renin activity & aldosterone | ↑ Renin, ↓ Aldosterone in primary Addison’s | | ✅ Autoantibody tests | Detect anti-adrenal antibodies in autoimmune cases | | ✅ Imaging (CT/MRI) | Evaluate adrenal gland size (atrophy, calcification, tumors, TB)
Pituitary MRI in secondary causes |
💊 Medical Management of Addison’s Disease
🎯 Goals of Treatment:
- Replace deficient hormones (cortisol and aldosterone)
- Manage electrolyte and fluid imbalances
- Educate for lifelong hormone therapy and stress-dose adjustments
🩺 1. Hormone Replacement Therapy (Lifelong):
| 💊 Drug | 📋 Purpose |
|---|---|
| Hydrocortisone (preferred) or prednisolone | Glucocorticoid replacement (2–3 divided doses/day) |
| Fludrocortisone acetate | Mineralocorticoid replacement (once daily) to maintain sodium and BP |
| Androgen replacement (optional in women) | DHEA may improve mood and libido |
🛑 2. Addisonian Crisis Management (Emergency):
- IV hydrocortisone 100 mg immediately
- Rapid IV fluids (normal saline with dextrose)
- Electrolyte correction: potassium-lowering therapy if needed
- Treat underlying cause (e.g., infection, stress, trauma)
🔎 3. Patient Education:
- Lifelong therapy is essential
- Increase steroid dose during stress, illness, surgery, or trauma
- Always carry medical alert ID and emergency steroid kit
- Regular follow-up for hormone levels and symptom monitoring
🛠️ Surgical Management
⚠️ Surgery is not a direct treatment for Addison’s disease but may be needed in certain conditions:
| 🛠️ Indication | 🔍 Reason |
|---|---|
| Adrenal tumors or cancer | Bilateral or unilateral adrenalectomy |
| Infection or hemorrhage requiring surgery | E.g., adrenal tuberculosis or bleeding adrenal gland |
| Secondary adrenal insufficiency due to pituitary tumor | Pituitary surgery to remove the tumor |
🛏️ Postoperative Considerations:
- Monitor for adrenal insufficiency post-op (especially if both glands removed)
- Lifelong glucocorticoid and mineralocorticoid replacement is mandatory after bilateral adrenalectomy
- Prevent Addisonian crisis by stress-dose steroid coverage before, during, and after surgery
🩺 NURSING MANAGEMENT OF ADDISON’S DISEASE
🎯 Nursing Goals:
- Prevent and manage Addisonian crisis
- Monitor and maintain fluid and electrolyte balance
- Administer lifelong hormone replacement therapy safely
- Educate patient and family for long-term self-care
- Monitor for complications and stress-dose adjustments
🗂️ I. Nursing Assessment
| 🔍 Assessment Focus | ✅ What to Monitor |
|---|---|
| Vital signs | Monitor for hypotension, weak pulse, dehydration, tachycardia |
| Neurological status | Assess for fatigue, confusion, lethargy (especially in crisis) |
| Skin | Look for hyperpigmentation, dryness |
| Electrolyte balance | Watch for hyponatremia, hyperkalemia, hypoglycemia |
| Fluid balance | Monitor I/O, daily weights, signs of dehydration |
| Medication history | Ask about steroid use, recent stress or illness |
| Signs of crisis | Vomiting, abdominal pain, extreme fatigue, shock signs |
📝 II. Common Nursing Diagnoses
- Deficient fluid volume related to aldosterone deficiency
- Risk for electrolyte imbalance related to decreased sodium and increased potassium
- Fatigue related to decreased cortisol and hypotension
- Knowledge deficit related to disease process and medication regimen
- Risk for Addisonian crisis related to stress, infection, or non-compliance
🧾 III. Nursing Interventions
🔹 During Addisonian Crisis (Emergency Care):
| 💡 Intervention | 🩺 Rationale |
|---|---|
| Administer IV hydrocortisone as ordered | Replaces deficient cortisol |
| Start IV fluids (Normal saline + 5% dextrose) | Corrects dehydration and hypoglycemia |
| Monitor cardiac rhythm, BP, and consciousness | Prevents complications from hyperkalemia and hypotension |
| Monitor blood glucose and electrolytes | Detects hypoglycemia, hyperkalemia, hyponatremia |
| Prepare for vasopressors if BP remains low despite fluids | Prevents circulatory collapse |
🔹 During Long-Term Management:
| 💊 Nursing Action | 💬 Purpose |
|---|---|
| Administer oral hydrocortisone and fludrocortisone | Maintain hormonal balance |
| Encourage liberal salt intake (unless contraindicated) | Compensates for sodium loss due to aldosterone deficiency |
| Educate patient about stress dosing | Prevents crisis during illness, injury, surgery |
| Monitor for signs of underdose (fatigue, low BP) and overdose (weight gain, edema) | Ensures proper dose adjustment |
| Encourage compliance with daily medication | Lifelong treatment is mandatory |
| Recommend wearing a medical alert bracelet | For emergencies and healthcare identification |
| Encourage regular follow-up and lab tests | Monitor hormone levels, electrolytes, kidney function |
🧑🏫 IV. Patient & Family Education
- 💊 Importance of lifelong medication adherence
- 📈 Recognize early signs of crisis: fatigue, nausea, low BP, fainting
- 💉 Teach how to use emergency injectable hydrocortisone (Solu-Cortef)
- 🚫 Never stop steroids abruptly
- 🤒 Increase steroid dose during illness, injury, surgery, emotional stress
- 📅 Schedule regular follow-ups with endocrinologist
- 🛡️ Encourage vaccination and infection prevention due to steroid immunosuppression
📊 V. Evaluation Criteria (Expected Outcomes)
- Patient maintains normal blood pressure, glucose, and electrolyte balance
- Demonstrates understanding of disease and treatment regimen
- Recognizes and responds appropriately to Addisonian crisis symptoms
- Takes medications regularly and correctly
- Maintains fluid balance and energy level
⚠️ COMPLICATIONS OF ADDISON’S DISEASE
❗ 1. Addisonian Crisis (Acute Adrenal Crisis)
Most serious and life-threatening complication
- Triggered by stress, infection, trauma, or sudden withdrawal of steroids
- Rapid drop in cortisol and aldosterone
- Symptoms:
- Severe hypotension or shock
- Dehydration
- Hypoglycemia
- Electrolyte imbalance (↑ potassium, ↓ sodium)
- Confusion or loss of consciousness
- Requires immediate IV hydrocortisone, fluids, and glucose
🧂 2. Chronic Electrolyte Imbalance
- Hyponatremia: fatigue, confusion, seizures
- Hyperkalemia: muscle weakness, arrhythmias
- Hypoglycemia: especially in children and fasting states
💓 3. Cardiovascular Instability
- Persistent low blood pressure
- Risk of cardiac arrhythmias from hyperkalemia or hypoglycemia
🧠 4. Neuropsychiatric Effects
- Mood changes, depression, irritability
- Lethargy, confusion, mental fog
🧬 5. Risk of Infection
- Long-term steroid therapy (for replacement) can suppress immunity
- Increased susceptibility to infections, delayed wound healing
📉 6. Adrenal Atrophy or Gland Destruction
- Especially in autoimmune cases or untreated TB
- Results in permanent adrenal insufficiency
🧷 KEY POINTS ON ADDISON’S DISEASE
✅ Definition: Primary adrenal insufficiency due to destruction or dysfunction of the adrenal cortex → ↓ cortisol + ↓ aldosterone
✅ Common Causes:
- Autoimmune adrenalitis (most common)
- Tuberculosis, infections
- Adrenalectomy or hemorrhage
- Genetic disorders (congenital hypoplasia)
✅ Types:
- Primary (Addison’s)
- Secondary (pituitary cause)
- Tertiary (hypothalamic or abrupt steroid withdrawal)
✅ Classic Symptoms:
- Fatigue, hypotension, hyperpigmentation, nausea, salt craving
- Electrolyte imbalance: ↓ Na⁺, ↑ K⁺, ↓ glucose
✅ Diagnosis:
- ↓ Morning cortisol, ↑ ACTH
- ACTH stimulation test
- Electrolyte panel
✅ Medical Management:
- Hydrocortisone + fludrocortisone (lifelong)
- IV steroids + fluids during crisis
✅ Nursing Focus:
- Prevent crisis, monitor vitals, educate on lifelong treatment and stress dosing
✅ Complications:
- Addisonian crisis, arrhythmias, infections, electrolyte imbalances
🌡️ CUSHING’S DISEASE
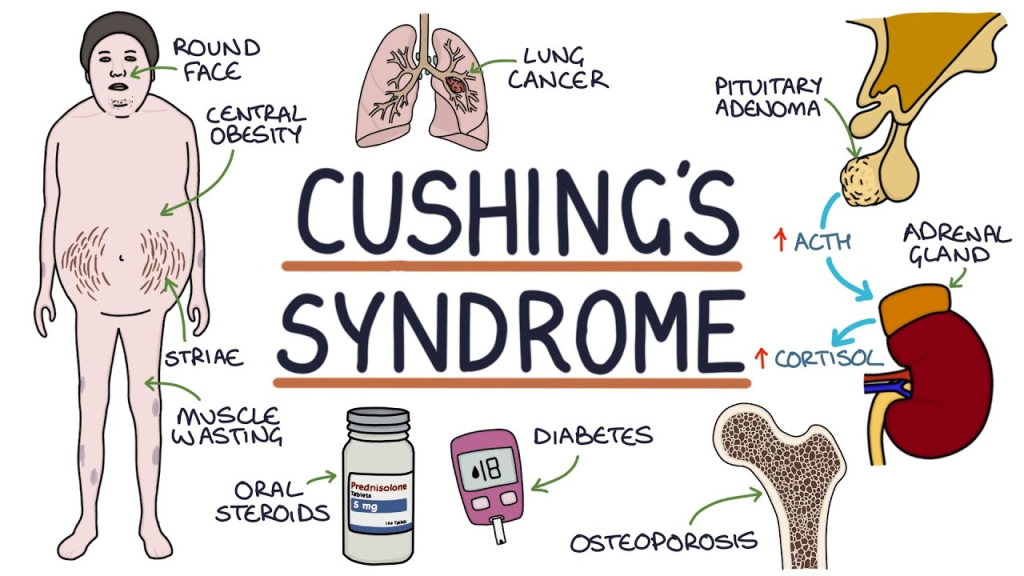
📌 Definition:
Cushing’s Disease is a specific form of Cushing’s Syndrome caused by a pituitary adenoma (benign tumor) that secretes excess adrenocorticotropic hormone (ACTH), leading to overstimulation of the adrenal glands and increased production of cortisol.
🧠 Cushing’s Syndrome is the general term for excess cortisol, regardless of cause.
Cushing’s Disease refers specifically to pituitary-origin ACTH overproduction.
🧬 Causes of Cushing’s Disease:
| 🎯 Cause | 📋 Description |
|---|---|
| Pituitary adenoma (ACTH-secreting) | Most common cause of Cushing’s disease (in ~70% of endogenous cases) |
| Ectopic ACTH-producing tumors (not Cushing’s Disease, but Cushing’s Syndrome) | Small cell lung cancer, pancreatic tumors |
| Exogenous corticosteroids (Cushing’s Syndrome) | Prolonged use of high-dose corticosteroids (e.g., prednisone, dexamethasone) |
| Adrenal tumors (adenoma/carcinoma) | Secrete cortisol directly (independent of ACTH) |
| CRH-secreting hypothalamic tumors | Rare cause, over-stimulates pituitary ACTH release |
⚠️ In Cushing’s Disease, the excess ACTH comes from the pituitary → adrenal hyperplasia → excess cortisol.
🏷️ Types of Cushing’s Syndrome (for differentiation):
| Type | Description |
|---|---|
| ACTH-dependent | Due to pituitary adenoma (Cushing’s disease) or ectopic ACTH production |
| ACTH-independent | Due to adrenal adenoma, carcinoma, or exogenous steroids |
| Iatrogenic (Exogenous) | From long-term corticosteroid therapy (most common overall) |
| Ectopic Cushing’s Syndrome | ACTH produced by non-pituitary tumors (e.g., lung cancer) |
🧬 Pathophysiology:
- ACTH-Secreting Pituitary Adenoma
A benign tumor in the anterior pituitary secretes excess adrenocorticotropic hormone (ACTH). - Adrenal Cortex Overstimulation
ACTH stimulates the adrenal cortex to overproduce cortisol (a glucocorticoid hormone). - Cortisol Excess Affects Multiple Systems:
- Carbohydrate metabolism → ↑ blood glucose
- Protein metabolism → muscle wasting, skin thinning
- Fat metabolism → abnormal fat distribution
- Mineralocorticoid effect → sodium retention, potassium loss
- Immune suppression → increased infection risk
- Feedback Mechanism Fails
High cortisol should normally suppress ACTH, but the pituitary tumor continues uncontrolled ACTH production.
🚨 Signs and Symptoms:
Mnemonic: “CUSHINGOID”
| 🔍 Category | 📝 Symptoms |
|---|---|
| C – Central obesity | Moon face, buffalo hump, truncal obesity |
| U – Unusual hair growth | Hirsutism (in women), acne |
| S – Skin changes | Thinning, bruising, purple striae on abdomen |
| H – Hypertension/Hyperglycemia | Due to cortisol’s mineralocorticoid and gluconeogenic effects |
| I – Infections | Increased susceptibility due to immune suppression |
| N – Neuropsychiatric | Depression, mood swings, memory issues |
| G – Glucose intolerance | May progress to diabetes mellitus |
| O – Osteoporosis | Bone resorption leads to fractures |
| I – Irregular menses/Impotence | Gonadal suppression in both sexes |
| D – Dorsocervical fat pad | “Buffalo hump” behind the neck |
🧪 Diagnosis of Cushing’s Disease:
🔹 1. Screening Tests (to confirm hypercortisolism):
| Test | Interpretation |
|---|---|
| 24-hour urinary free cortisol | ↑ Elevated cortisol |
| Late-night salivary cortisol | ↑ Loss of circadian rhythm |
| Low-dose dexamethasone suppression test (LDDST) | No suppression of cortisol = abnormal |
🔹 2. Confirm ACTH-Dependent Source:
| Test | Interpretation |
|---|---|
| Plasma ACTH level | ↑ High in Cushing’s Disease |
| High-dose dexamethasone suppression test | Cortisol suppression suggests pituitary origin (vs ectopic) |
🔹 3. Imaging:
| Test | Purpose |
|---|---|
| MRI of the pituitary gland | Detects pituitary adenoma |
| CT of adrenal glands | Rules out adrenal tumors if ACTH is low |
| Inferior petrosal sinus sampling (IPSS) | Gold standard to confirm pituitary source of ACTH (especially if MRI is inconclusive) |
💊 I. MEDICAL MANAGEMENT
🎯 Goals of Treatment:
- Reduce cortisol levels
- Treat the underlying pituitary adenoma
- Manage metabolic and systemic complications
- Prepare for or avoid surgery if needed
✅ 1. Medications to Reduce Cortisol Production:
| Drug | Class | Purpose |
|---|---|---|
| Ketoconazole | Antifungal (inhibits steroid synthesis) | Blocks cortisol production in adrenal glands |
| Metyrapone | Adrenal enzyme inhibitor | Decreases cortisol levels |
| Mitotane | Adrenolytic agent | Used in adrenal carcinoma; suppresses adrenal function |
| Pasireotide | Somatostatin analog | Inhibits ACTH secretion from pituitary tumors |
| Cabergoline | Dopamine agonist | May reduce ACTH levels in some pituitary tumors |
🔄 Used when surgery is contraindicated, as preoperative preparation, or for residual/recurrent disease
✅ 2. Supportive Treatment for Complications:
- Antihypertensives – for high blood pressure
- Antidiabetic drugs – for glucose control
- Bisphosphonates or calcium/vitamin D – for osteoporosis
- Antidepressants or anxiolytics – for mood disturbances
- High-protein, low-carb, low-sodium diet
🛠️ II. SURGICAL MANAGEMENT
🎯 Surgery is the definitive treatment for Cushing’s Disease (pituitary cause of hypercortisolism)
🔪 1. Transsphenoidal Surgery (TSS):
- Most common surgical treatment
- Endoscopic removal of pituitary adenoma via the nose/sphenoid sinus
- Preferred for small, localized tumors
- Success rate: ~80–90% with low recurrence
🔪 2. Adrenalectomy (Unilateral/Bilateral):
| Type | Indication |
|---|---|
| Unilateral | If adrenal tumor is source of excess cortisol |
| Bilateral | In refractory Cushing’s disease or failed pituitary surgery |
❗ Requires lifelong hormone replacement therapy (glucocorticoids & mineralocorticoids)
🔬 3. Radiation Therapy:
- Used when:
- Surgery is not possible
- Tumor recurs or is incompletely removed
- Stereotactic radiosurgery (e.g., Gamma Knife) may be used
- Takes months to show full effect
🩺 Post-Surgical Considerations:
| Focus Area | Nursing Implication |
|---|---|
| Adrenal insufficiency | Common after successful surgery → monitor for hypotension, weakness, hyponatremia |
| Temporary hormone replacement | Hydrocortisone post-op, taper gradually |
| Monitor cortisol & ACTH levels | To assess surgical success and guide therapy |
| Watch for infection or CSF leak (TSS) | Especially in nasal surgeries |
| Educate about lifelong hormone monitoring | Especially after adrenalectomy |
🩺 NURSING MANAGEMENT OF CUSHING’S DISEASE
🎯 Goals of Nursing Care:
- Monitor and stabilize vital signs and lab values
- Reduce cortisol-related complications
- Support the patient pre- and post-operatively
- Promote emotional and physical well-being
- Educate the patient for lifelong health monitoring
🗂️ I. Nursing Assessment
| 🔍 Assessment Area | ✅ Focus |
|---|---|
| Vital signs | Monitor for hypertension, tachycardia, fever (infection risk) |
| Skin integrity | Check for fragile skin, bruises, delayed wound healing |
| Muscle strength | Assess for muscle wasting, fatigue |
| Mental status | Observe for mood swings, depression, anxiety |
| Body measurements | Monitor for weight gain, central obesity, moon face |
| Glucose monitoring | Check for hyperglycemia and signs of diabetes |
| Bone pain or deformities | Screen for osteoporosis-related complications |
📝 II. Common Nursing Diagnoses
- Risk for infection related to immunosuppression
- Imbalanced nutrition: More than body requirements related to cortisol excess
- Disturbed body image related to physical changes
- Impaired skin integrity due to thinning and bruising
- Risk for injury related to osteoporosis and muscle weakness
- Knowledge deficit related to disease process and lifelong management
🧾 III. Nursing Interventions
🔹 Preoperative Care (if surgery planned):
| 🩺 Intervention | 💡 Rationale |
|---|---|
| Administer prescribed medications (e.g., ketoconazole) | To reduce cortisol pre-op |
| Maintain fluid and electrolyte balance | Prevent complications from hypertension or edema |
| Monitor glucose levels and treat hyperglycemia | Cortisol increases blood sugar |
| Educate on surgical procedure and recovery | Reduce anxiety and promote cooperation |
🔹 Postoperative Care (e.g., after transsphenoidal surgery):
| 🩺 Intervention | 💡 Rationale |
|---|---|
| Monitor for adrenal insufficiency | Sudden cortisol drop may lead to hypotension, weakness, nausea |
| Administer IV hydrocortisone as prescribed | Prevent acute adrenal crisis |
| Monitor fluid balance and daily weight | Detect early signs of fluid overload or dehydration |
| Monitor for nasal drainage or CSF leak | Post-transsphenoidal surgery complication |
| Encourage oral hygiene and avoid nose blowing | Prevent pressure at surgical site |
| Watch for infection signs | Immunosuppression increases risk |
🔹 Long-Term Care and Patient Education:
| 🧠 Education Topic | 🧾 Key Points |
|---|---|
| Medication adherence | Continue hormone therapy as prescribed |
| Follow-up appointments | Monitor cortisol, ACTH, glucose, and bone health |
| Diet and exercise | Encourage high-protein, low-sodium, low-carb diet |
| Skin care | Use gentle soaps, avoid trauma to fragile skin |
| Stress management | Cortisol affects mood—support mental health |
| Recognition of signs of recurrence or adrenal insufficiency | Fatigue, hypotension, nausea, dizziness |
📊 IV. Evaluation Criteria (Expected Outcomes):
- Cortisol levels return to or maintain within normal range
- Patient is free from infections, skin breakdown, and injuries
- Body weight and BP are controlled
- Patient verbalizes understanding of disease, treatment, and follow-up care
- Patient demonstrates positive coping and adjustment to body image changes
⚠️ COMPLICATIONS OF CUSHING’S DISEASE
Cushing’s disease causes widespread metabolic and systemic changes. If left untreated or poorly managed, it can lead to serious complications:
🦴 1. Osteoporosis & Pathological Fractures
- Cortisol inhibits bone formation and enhances bone resorption
- High risk of vertebral compression fractures, hip fractures, and bone pain
💓 2. Hypertension and Cardiovascular Disease
- Cortisol has mineralocorticoid effects → sodium and water retention
- Leads to persistent high blood pressure, increasing risk of:
- Stroke
- Heart failure
- Myocardial infarction
🍬 3. Hyperglycemia and Diabetes Mellitus
- Cortisol increases gluconeogenesis → elevated blood glucose
- Can lead to insulin resistance and type 2 diabetes
🦠 4. Immunosuppression and Increased Infections
- Cortisol suppresses the immune response
- Increased risk of skin infections, respiratory infections, delayed wound healing
🧠 5. Neuropsychiatric Issues
- Mood swings, depression, insomnia, cognitive decline
- Severe cases may involve psychosis or suicidal ideation
🧍 6. Body Image Disturbance and Social Withdrawal
- Due to moon face, central obesity, striae, and hirsutism
- Can lead to low self-esteem, anxiety, and social isolation
💊 7. Adrenal Insufficiency (Post-treatment)
- After surgery or long-term medical treatment, adrenal glands may become suppressed
- Sudden withdrawal of cortisol can cause adrenal crisis
🧷 KEY POINTS ON CUSHING’S DISEASE
✅ Definition: Endogenous hypercortisolism caused by a pituitary adenoma that secretes excess ACTH
✅ Distinction:
- Cushing’s Syndrome = cortisol excess (any cause)
- Cushing’s Disease = pituitary-based ACTH overproduction
✅ Classic Signs:
- Moon face, buffalo hump, truncal obesity
- Purple striae, fragile skin, easy bruising
- Hypertension, hyperglycemia, hirsutism
- Mood swings, menstrual irregularities
✅ Diagnosis:
- ↑ Cortisol (saliva, urine, or serum)
- ↑ ACTH
- MRI of pituitary
- Dexamethasone suppression test
✅ Treatment:
- Transsphenoidal surgery (first-line)
- Medical therapy (ketoconazole, metyrapone)
- Hormonal and metabolic monitoring
✅ Nursing Role:
- Monitor vitals, glucose, wound healing
- Support medication and treatment adherence
- Educate on body image coping, long-term follow-up
✅ Complications:
- Diabetes, hypertension, fractures, infections, psychiatric issues, adrenal insufficiency
🧬 CONGENITAL ADRENAL HYPERPLASIA (CAH)
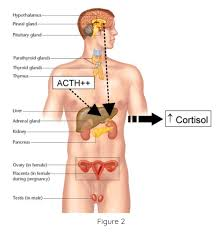
📌 Definition:
Congenital Adrenal Hyperplasia (CAH) is a group of inherited autosomal recessive disorders characterized by enzyme deficiencies in the adrenal cortex that impair the synthesis of cortisol, and sometimes aldosterone, leading to excess androgen production.
🧠 CAH leads to adrenal hyperplasia due to increased ACTH stimulation, and can result in ambiguous genitalia, salt-wasting crisis, or early puberty, depending on the enzyme involved.
🧬 Causes:
| 🔍 Cause | 📋 Description |
|---|---|
| Genetic Mutation | CAH is caused by mutations in genes responsible for enzymes that synthesize adrenal hormones (mainly 21-hydroxylase deficiency) |
| Autosomal Recessive Inheritance | Both parents must carry a mutated gene |
| Enzyme Deficiencies | These impair cortisol (and/or aldosterone) synthesis, leading to: |
- Reduced negative feedback
- ↑ ACTH from pituitary
- Adrenal hyperplasia
- Excess androgen production (testosterone-like hormones)
🏷️ Types of CAH (Based on Enzyme Deficiency):
| 🔬 Type | % Cases | Clinical Features |
|---|---|---|
| 1. 21-hydroxylase deficiency (most common) | ~90–95% | ↓ Cortisol ± ↓ Aldosterone → ↑ ACTH → ↑ Androgens |
| – Classic Salt-Wasting Type | Severe | Cortisol & aldosterone both deficient → vomiting, dehydration, shock in neonates |
| – Classic Simple Virilizing Type | Moderate | Cortisol deficient; aldosterone normal → ambiguous genitalia in females; early puberty in males |
| – Non-Classic Type | Mild/Late onset | Presents in childhood/adolescence with hirsutism, acne, irregular menses |
| 2. 11β-hydroxylase deficiency | ~5–8% | ↑ 11-deoxycorticosterone (mineralocorticoid effect) → hypertension + virilization |
| 3. 17α-hydroxylase deficiency | Rare | ↓ Sex steroids → sexual infantilism and hypertension |
| 4. 3β-hydroxysteroid dehydrogenase deficiency | Very rare | Defects in all three adrenal pathways (glucocorticoid, mineralocorticoid, sex steroids) |
🧬 Pathophysiology of CAH:
- 🧪 Genetic mutation causes a deficiency in enzymes required for the synthesis of:
- Cortisol (main glucocorticoid)
- Aldosterone (mineralocorticoid, in some types)
- With a compensatory increase in ACTH due to lack of cortisol feedback
- ⚠️ ACTH overstimulation → adrenal cortex hyperplasia (enlargement)
- ⚙️ The hormonal precursors accumulate and get diverted into the androgen (testosterone) pathway
- 🔄 Depending on the type of CAH:
- Cortisol deficiency leads to poor stress response
- Aldosterone deficiency leads to salt wasting, dehydration, hyperkalemia
- Androgen excess causes virilization of female genitalia and early puberty in males
- Mineralocorticoid excess (e.g., in 11β-hydroxylase deficiency) may cause hypertension
🚨 Signs and Symptoms of CAH
Vary depending on type (classic salt-wasting, simple virilizing, or non-classic)
👶 In Neonates (Classic CAH):
| 👧 Female Infants | 👦 Male Infants |
|---|---|
| Ambiguous genitalia (clitoral enlargement, fused labia) | Normal genitalia at birth (may be missed) |
| Internal female organs are normal | Symptoms develop later |
| Salt-wasting crisis within 1–2 weeks if aldosterone is deficient (vomiting, dehydration, hypotension, shock) |
🧒 In Children and Adolescents:
- Early puberty (precocious puberty)
- Rapid growth in early childhood, but premature closure of epiphyses → short adult height
- Acne, oily skin
- Early development of pubic/axillary hair (pubarche)
- Irregular menstruation in females
- Behavioral changes, aggression (linked to high androgens)
👩 In Adults (Non-Classic CAH):
- Hirsutism (excess facial/body hair)
- Acne, oily skin
- Menstrual irregularities or amenorrhea
- Infertility
- Polycystic ovary-like symptoms
🧪 Diagnosis of CAH
Early diagnosis is critical, especially in salt-wasting forms to prevent crisis.
✅ Neonatal Screening:
- Done within 48–72 hours of birth
- Measures 17-hydroxyprogesterone (17-OHP): elevated in 21-hydroxylase deficiency
✅ Hormonal Blood Tests:
| Test | Expected Findings |
|---|---|
| 17-hydroxyprogesterone | ↑ High in most forms of CAH |
| ACTH | ↑ Elevated |
| Cortisol | ↓ Low or normal |
| Aldosterone | ↓ Low in salt-wasting type; ↑ in 11β-hydroxylase deficiency |
| Renin activity | ↑ High in salt-wasting CAH |
| Androgens (DHEA, testosterone) | ↑ Elevated in virilizing forms |
🧬 Additional Tests:
- Electrolyte panel:
- ↓ Sodium
- ↑ Potassium (in salt-wasting type)
- Acidosis
- Pelvic ultrasound (in females):
- To detect internal reproductive organs
- Genetic testing:
- For mutation identification and family counseling
💊 I. MEDICAL MANAGEMENT
🎯 Goals of Treatment:
- Replace deficient hormones (cortisol and/or aldosterone)
- Suppress excess ACTH to prevent adrenal hyperplasia and androgen excess
- Prevent or treat salt-wasting crises
- Promote normal growth and development
✅ 1. Hormone Replacement Therapy
| Medication | Purpose |
|---|---|
| Hydrocortisone (oral) | Replaces cortisol and suppresses ACTH (first-line in children) |
| Prednisolone or Dexamethasone | Used in older children or adults (longer acting) |
| Fludrocortisone | Replaces aldosterone in salt-wasting CAH |
| Salt supplements (NaCl) | Given in infants with salt-wasting form, especially under 6 months |
✅ 2. Monitoring and Adjustments
| Parameter | Frequency |
|---|---|
| Growth and weight | Every 3 months |
| Serum electrolytes | Every 3–6 months in salt-wasting types |
| 17-OHP levels | Used to monitor therapy response |
| Bone age X-ray | To assess skeletal maturation |
| Blood pressure and glucose | Routine monitoring to avoid over-treatment |
✅ 3. Treatment of Acute Adrenal Crisis
Emergency situation—especially in undiagnosed neonates or poor compliance
Signs: Vomiting, dehydration, hypotension, shock, hypoglycemia, hyperkalemia
Treatment:
- IV hydrocortisone
- IV normal saline + 5% dextrose
- Correct electrolyte imbalances
- Hospitalization and intensive monitoring
🛠️ II. SURGICAL MANAGEMENT
🎯 Surgery is not always required but may be necessary in some cases for genital reconstruction or associated complications.
✅ 1. Genital Reconstruction Surgery (in Female Infants):
| Indication | Description |
|---|---|
| Ambiguous genitalia | Surgery may be considered to correct clitoral enlargement, fused labia, and to create a functional vagina |
| Timing | Ideally between 6–12 months of age, depending on family preference and expert team recommendations |
| Types of Surgery | Clitoroplasty, vaginoplasty, or combined genitoplasty |
🔔 Surgical decisions must involve parents, endocrinologist, surgeon, and psychologist, respecting future gender identity and individual rights.
✅ 2. Other Surgical Indications:
- Hernia repair (common in infants)
- Management of complications like urinary obstruction or infections
🔍 Post-Surgical Care:
- Pain management, wound care
- Prevention of infection
- Emotional support for family
- Continued hormonal therapy
- Long-term follow-up with urology, endocrinology, and psychology
🩺 NURSING MANAGEMENT OF CONGENITAL ADRENAL HYPERPLASIA (CAH)
🎯 Nursing Goals:
- Maintain fluid and electrolyte balance
- Prevent and manage adrenal crisis
- Ensure compliance with lifelong hormone therapy
- Support growth and development
- Provide emotional and psychosocial support to the child and family
- Educate for long-term management and follow-up
🗂️ I. Nursing Assessment
| 🔍 Area | ✅ Focus Points |
|---|---|
| Vital signs | Monitor for hypotension, dehydration, or signs of shock |
| Hydration status | Assess intake/output, mucous membranes, fontanelles (infants) |
| Electrolyte levels | Watch for hyponatremia, hyperkalemia, and hypoglycemia |
| Growth monitoring | Track height, weight, and bone age regularly |
| Developmental milestones | Identify any delays in motor, social, or cognitive areas |
| Genital appearance | Observe and record findings (in females); refer for surgical opinion if needed |
| Medication adherence | Assess family’s understanding and compliance with hormonal therapy |
📝 II. Common Nursing Diagnoses
- Risk for fluid volume deficit related to salt-wasting form
- Imbalanced nutrition related to increased metabolic needs
- Risk for delayed growth and development related to hormonal imbalance
- Risk for adrenal crisis during illness, stress, or surgery
- Disturbed body image (in older children/adolescents with ambiguous genitalia or short stature)
- Deficient knowledge related to disease process, treatment, and follow-up needs
🧾 III. Nursing Interventions
🔹 A. Acute Care (During Salt-Wasting Crisis or Emergency):
| 🛑 Intervention | 🎯 Purpose |
|---|---|
| Administer IV hydrocortisone, fluids, and dextrose as prescribed | Stabilize hormones and treat shock |
| Monitor electrolytes, glucose, and BP | Detect life-threatening imbalances |
| Maintain strict I&O records | Guide fluid replacement |
| Observe for signs of vomiting, diarrhea, poor feeding | Early signs of crisis in infants |
🔹 B. Routine/Long-Term Care:
| 🩺 Intervention | 🎯 Purpose |
|---|---|
| Administer oral hydrocortisone and fludrocortisone as prescribed | Maintain hormone balance |
| Encourage salt supplementation in infants if prescribed | Prevent hyponatremia |
| Monitor growth charts and development regularly | Detect stunting or early puberty |
| Provide age-appropriate play and stimulation | Promote normal development |
| Involve multidisciplinary team (endocrinologist, surgeon, psychologist) | Holistic care planning |
| Support child and family emotionally | Address parental guilt, confusion, or anxiety |
| Teach about stress dosing of steroids | Prepare for illness, surgery, trauma situations |
| Ensure access to medical ID tag and emergency steroid kit | For safety during emergencies |
🔹 C. Family Education:
| 📘 Topic | 📌 Education Content |
|---|---|
| Disease process | Explain genetic nature, hormone imbalance, and treatment goals |
| Medication adherence | Emphasize need for lifelong treatment and exact timing |
| Emergency care | Teach signs of adrenal crisis and when to seek immediate help |
| Gender development and surgical decisions | Respect family’s decisions and support with information |
| Follow-up importance | Regular labs, growth assessments, imaging as needed |
📊 IV. Evaluation Criteria (Expected Outcomes):
- Child maintains normal hydration, BP, and electrolyte levels
- Child demonstrates age-appropriate growth and development
- Family demonstrates understanding of medication schedule and emergency plan
- No episodes of adrenal crisis or hospitalization due to poor compliance
- Emotional and psychological needs of child and parents are addressed appropriately
⚠️ COMPLICATIONS OF CONGENITAL ADRENAL HYPERPLASIA (CAH)
🧪 1. Adrenal Crisis (Life-Threatening Emergency)
- Often occurs during illness, surgery, injury, or stress
- Triggered by inadequate cortisol levels
- Symptoms:
- Severe vomiting, diarrhea
- Dehydration, hypotension, shock
- Hypoglycemia, hyponatremia, hyperkalemia
- Requires urgent IV hydrocortisone, fluids, and electrolyte correction
📉 2. Growth Abnormalities
- Tall stature in early childhood, followed by short adult height due to early epiphyseal fusion
- Stunted growth in undertreated or over-treated cases
🧠 3. Cognitive and Developmental Delays
- If not treated early, CAH may lead to delayed milestones, learning difficulties, or poor school performance
- Especially in salt-wasting crisis survivors with delayed diagnosis
👧 4. Ambiguous Genitalia & Psychosocial Impact (Females)
- Clitoral enlargement, fused labia may lead to:
- Parental distress
- Body image issues
- Gender identity concerns in adolescence
- Need for psychosocial counseling and support
🧍 5. Fertility Issues
- Common in untreated or poorly controlled CAH, especially in females
- Menstrual irregularities, anovulation, and PCOS-like features
💊 6. Medication Side Effects (from Long-Term Steroid Use)
- Cushingoid features if over-treated: weight gain, moon face, glucose intolerance
- Osteoporosis, hypertension, and glucose abnormalities with long-term high-dose steroid use
🧷 KEY POINTS ON CONGENITAL ADRENAL HYPERPLASIA (CAH)
✅ Definition: CAH is a group of inherited enzyme deficiencies affecting cortisol (and sometimes aldosterone) production → adrenal hyperplasia + excess androgens
✅ Most common cause: 21-hydroxylase deficiency
✅ Types:
- Classic Salt-Wasting CAH – cortisol and aldosterone deficiency
- Simple Virilizing CAH – cortisol deficiency, normal aldosterone
- Non-Classic CAH – mild form, appears later in life
✅ Pathophysiology:
- ↓ Cortisol → ↑ ACTH → adrenal hyperplasia → ↑ androgen production
- May result in ambiguous genitalia, precocious puberty, or infertility
✅ Symptoms:
- Salt-wasting crisis, ambiguous genitalia in females, early virilization, rapid growth, short adult height
✅ Diagnosis:
- ↑ 17-OHP, abnormal electrolytes, ACTH, androgens
- Confirm with newborn screening, hormone tests, and genetic testing
✅ Treatment:
- Lifelong hydrocortisone, fludrocortisone, and salt supplementation
- Surgical correction of genitalia (when needed)
- Emergency care with stress dosing of steroids during illness
✅ Nursing Role:
- Monitor growth, electrolytes, adherence
- Educate on medications, stress dosing, and emergency response
- Provide family counseling and emotional support
✅ Complications:
- Adrenal crisis, growth issues, infertility, psychosocial problems, and steroid side effects
⚡ PHEOCHROMOCYTOMA
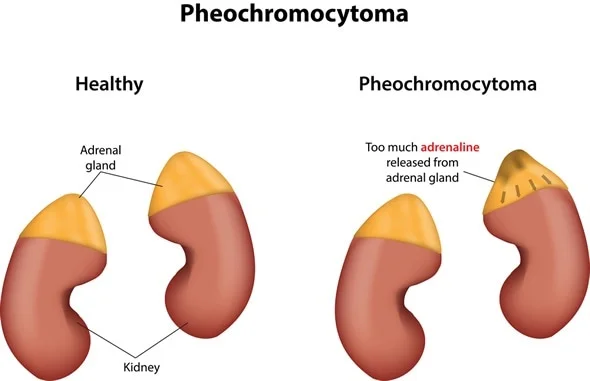
📌 Definition:
Pheochromocytoma is a rare, usually benign tumor that arises from the chromaffin cells of the adrenal medulla and produces excess catecholamines—primarily epinephrine (adrenaline) and norepinephrine (noradrenaline).
🔺 These hormones cause sudden or sustained episodes of hypertension, palpitations, sweating, and other fight-or-flight symptoms.
🧬 Causes:
| 🔍 Cause | 📋 Description |
|---|---|
| Genetic mutations (familial forms) | 25–30% of cases are hereditary; mutations in genes such as RET, VHL, NF1, and SDH |
| Sporadic (non-hereditary) | Most common in adults, no family history |
| Associated syndromes | – Multiple Endocrine Neoplasia type 2 (MEN 2A & 2B) |
- Von Hippel-Lindau disease
- Neurofibromatosis type 1 (NF1) | | Tumors outside adrenal gland (paraganglioma) | Arise from extra-adrenal chromaffin tissue; may also secrete catecholamines |
🏷️ Types of Pheochromocytoma:
| Type | Description |
|---|---|
| Adrenal Pheochromocytoma | Arises from the adrenal medulla (most common form) |
| Extra-adrenal Pheochromocytoma (Paraganglioma) | Arises from sympathetic ganglia outside the adrenal glands—may be abdominal, thoracic, pelvic |
| Benign Pheochromocytoma | Non-cancerous and confined to adrenal gland (majority of cases) |
| Malignant Pheochromocytoma | Rare; defined by metastasis to organs like bones, lungs, liver, or lymph nodes |
| Familial Pheochromocytoma | Occurs as part of hereditary syndromes (MEN 2, VHL, NF1); often bilateral or multiple tumors |
🧬 Pathophysiology of Pheochromocytoma:
- Tumor develops in chromaffin cells of the adrenal medulla or extra-adrenal sympathetic tissue (paraganglia).
- These tumor cells overproduce catecholamines:
- Epinephrine (adrenaline)
- Norepinephrine (noradrenaline)
- Occasionally dopamine
- Excess catecholamines stimulate alpha- and beta-adrenergic receptors:
- ⬆️ Vasoconstriction → hypertension
- ⬆️ Heart rate and contractility → tachycardia
- ⬆️ Glycogenolysis → hyperglycemia
- Intermittent or sustained secretion causes episodic or persistent hypertensive crises, potentially life-threatening if untreated.
- High catecholamines can damage heart, kidneys, brain, and increase the risk of stroke or MI.
🚨 Signs and Symptoms of Pheochromocytoma:
Classic “Rule of 10s” (old teaching, less used now):
10% bilateral, 10% malignant, 10% extra-adrenal, 10% familial (now known to be ~30% familial)
📌 Classic Triad of symptoms:
- Headache (severe, pounding)
- Palpitations (with tachycardia)
- Sweating (diaphoresis)
🧠 Other Common Symptoms:
| System | Symptoms |
|---|---|
| Cardiovascular | – Sudden or sustained hypertension (may be episodic) |
- Tachycardia, palpitations
- Orthostatic hypotension (due to volume depletion)
- Chest pain, arrhythmias | | Neurological | – Headache
- Anxiety or panic attack-like symptoms
- Tremors
- Visual disturbances | | Metabolic | – Weight loss
- Heat intolerance
- Hyperglycemia | | Gastrointestinal | – Nausea, vomiting
- Abdominal or flank pain (tumor mass effect) | | Other | – Fatigue
- Pallor during attacks
- Feeling of impending doom |
⚠️ Symptoms may be triggered by stress, surgery, anesthesia, exercise, or medications (e.g., beta-blockers without alpha-blockade)
🧪 Diagnosis of Pheochromocytoma:
✅ Biochemical Tests (first-line):
| Test | Interpretation |
|---|---|
| Plasma free metanephrines | Highly sensitive; elevated in most cases |
| 24-hour urine catecholamines & metanephrines | Confirms diagnosis; includes norepinephrine, epinephrine, and their metabolites |
| Clonidine suppression test | Clonidine should suppress catecholamines in non-tumor cases; no suppression = positive |
✅ Imaging Studies (to localize tumor):
| Test | Purpose |
|---|---|
| CT scan or MRI of abdomen/pelvis | Detects adrenal or extra-adrenal tumors |
| MIBG (metaiodobenzylguanidine) scan | Nuclear medicine test to locate pheochromocytomas/paragangliomas, especially metastatic or extra-adrenal tumors |
| PET scan | Useful in malignant cases or when MIBG is inconclusive |
| Genetic testing | For familial syndromes (MEN 2, VHL, NF1, SDH mutations) |
💊 I. MEDICAL MANAGEMENT
🎯 Goal:
Stabilize the patient’s blood pressure and heart rate, prevent hypertensive crisis, and prepare for safe tumor removal.
✅ 1. Preoperative Medical Stabilization (Essential Step)
| Drug Type | Drug Name | Purpose |
|---|---|---|
| Alpha-blockers (first-line) | Phenoxybenzamine (long-acting) | |
| or Prazosin, Doxazosin | – Blocks vasoconstriction |
- Controls hypertension
- Initiated 7–14 days before surgery | | Beta-blockers | Propranolol, Atenolol | – Added after adequate alpha blockade
- Controls tachycardia, arrhythmias
⚠️ Never start beta-blockers before alpha-blockers (can worsen hypertensive crisis) | | Calcium channel blockers (optional) | Nifedipine, Amlodipine | Alternative or adjunct for BP control | | Volume expansion (IV fluids, salt) | – | Prevent post-op hypotension from vasodilation and depleted volume |
✅ 2. Management of Hypertensive Crisis (Emergency)
| Treatment | Purpose |
|---|---|
| IV Nitroprusside, Phentolamine, Nicardipine | Rapid BP control |
| IV Esmolol or Labetalol | Heart rate control (after alpha-blockade) |
| IV fluids | Correct volume depletion |
| ICU monitoring | For severe crises or arrhythmias |
🛠️ II. SURGICAL MANAGEMENT
🎯 Definitive Treatment: Surgical removal of the tumor (adrenalectomy)
✅ 1. Laparoscopic Adrenalectomy (Preferred for localized tumors)
- Minimally invasive
- Performed after optimal BP and HR control
- Most common approach for benign unilateral pheochromocytomas
✅ 2. Open Adrenalectomy
- Indicated in:
- Large tumors
- Suspected malignancy
- Bilateral tumors
- Extra-adrenal paragangliomas
✅ 3. Bilateral Adrenalectomy (in rare cases)
- Required in:
- Bilateral tumors (e.g., familial syndromes)
- Refractory or malignant cases
Requires lifelong glucocorticoid and mineralocorticoid replacement therapy (Addisonian management)
🩺 Postoperative Monitoring
| Focus Area | Action |
|---|---|
| BP & HR | Watch for hypotension after catecholamine drop |
| Blood glucose | Hypoglycemia may occur (due to sudden cortisol drop) |
| Adrenal insufficiency signs | Especially after bilateral adrenalectomy |
| Urine output & electrolytes | Monitor volume status |
| Pain & infection control | Standard post-op care |
🩺 NURSING MANAGEMENT OF PHEOCHROMOCYTOMA
🎯 Nursing Goals:
- Maintain hemodynamic stability
- Prevent hypertensive crisis or complications
- Ensure safe preparation and recovery from surgery
- Educate patient on lifelong monitoring and emergency preparedness
- Support emotional well-being and medication adherence
🗂️ I. Nursing Assessment
| 🔍 Assessment Area | ✅ Focus |
|---|---|
| Vital signs | Monitor BP, HR frequently (watch for sudden spikes) |
| Neurological status | Check for headache, restlessness, visual changes |
| Cardiac status | Assess for palpitations, arrhythmias, chest pain |
| Respiratory status | Observe for tachypnea or dyspnea |
| Fluid status | Monitor I&O, daily weight, signs of dehydration or volume overload |
| Blood glucose levels | Catecholamine excess can cause hyperglycemia, post-op hypoglycemia |
| Anxiety levels | Due to episodic attacks and fear of crisis |
📝 II. Common Nursing Diagnoses
- Risk for unstable blood pressure related to excess catecholamines
- Risk for decreased cardiac output related to arrhythmias and tachycardia
- Anxiety related to unpredictable attacks and fear of complications
- Deficient knowledge related to disease, medications, and post-op care
- Risk for injury due to sudden hypotension post-operatively
🧾 III. Nursing Interventions
🔹 A. Preoperative Phase:
| 🩺 Intervention | 💡 Purpose |
|---|---|
| Administer alpha-blockers (e.g., phenoxybenzamine) | To control BP and reduce tumor sensitivity |
| Add beta-blockers only after alpha-blockade | To manage tachycardia and prevent crisis |
| Provide high-sodium diet and fluid intake | To expand volume and prevent post-op hypotension |
| Monitor for headache, flushing, sweating, palpitations | Signs of catecholamine surges |
| Avoid emotional stress, pain, strenuous activity | These can trigger hypertensive episodes |
| Educate about surgery and post-op expectations | Reduce anxiety and ensure compliance |
🔹 B. Intraoperative/Postoperative Phase:
| 🩺 Post-Op Care | 💡 Action |
|---|---|
| Monitor BP and HR closely | Risk of sudden hypotension post tumor removal |
| Watch for adrenal insufficiency signs | Especially after bilateral adrenalectomy (fatigue, low BP, vomiting) |
| Administer IV fluids as ordered | To maintain perfusion and prevent hypotension |
| Monitor glucose levels | Risk of hypoglycemia after catecholamine drop |
| Observe for urine output | To assess kidney perfusion |
| Provide pain relief and emotional support | Aid recovery and comfort |
🔹 C. Long-Term & Discharge Education:
| 📘 Topic | 🧾 Teaching Points |
|---|---|
| Medication adherence | Continue any prescribed alpha-blockers until advised to stop |
| Recognize recurrence signs | Headache, palpitations, high BP |
| Follow-up appointments | For lab tests (metanephrines), imaging, genetic testing |
| Family screening | If hereditary type suspected |
| Lifelong steroids | If bilateral adrenalectomy was performed |
| Emergency awareness | Teach patient to carry medical alert ID and steroid emergency kit (if needed) |
📊 IV. Evaluation Criteria (Expected Outcomes):
- Patient maintains stable BP and HR
- Free from hypertensive or hypotensive episodes
- Verbalizes understanding of medications and disease
- Recovers safely from surgery without complications
- Demonstrates positive coping and confidence in managing health
⚠️ COMPLICATIONS OF PHEOCHROMOCYTOMA
Pheochromocytoma can cause life-threatening cardiovascular and metabolic emergencies if not properly managed.
💓 1. Hypertensive Crisis
- Sudden, severe rise in BP due to catecholamine surge
- May be spontaneous or triggered by:
- Stress, surgery, anesthesia
- Certain drugs (e.g., beta-blockers without alpha-blockade)
🛑 Can lead to:
- Stroke
- Myocardial infarction (heart attack)
- Heart failure
- Aortic dissection
🧠 2. Neurological Complications
- Stroke (hemorrhagic or ischemic) due to extreme hypertension
- Seizures from hypertensive encephalopathy
- Visual disturbances or blindness (retinopathy)
💓 3. Arrhythmias and Tachycardia
- Caused by catecholamine overstimulation of β1-receptors
- May lead to ventricular arrhythmias or sudden cardiac death
🫀 4. Cardiomyopathy (Catecholamine-Induced)
- Known as stress-induced (Takotsubo) cardiomyopathy
- Left ventricular dysfunction due to prolonged catecholamine toxicity
- Can mimic acute coronary syndrome
💧 5. Postoperative Hypotension & Hypoglycemia
- Sudden drop in catecholamines after tumor removal → hypotension
- Sudden cortisol/epinephrine reduction → hypoglycemia
🧬 6. Malignant Transformation or Recurrence
- Rare, but malignant pheochromocytomas can metastasize
- Requires lifelong surveillance, especially in familial types
🧷 KEY POINTS ON PHEOCHROMOCYTOMA
✅ Definition: Tumor of adrenal medulla or sympathetic ganglia that secretes excess catecholamines (epinephrine, norepinephrine)
✅ Classic Triad:
- Headache
- Palpitations
- Sweating
✅ Other Signs:
- Paroxysmal or sustained hypertension
- Tremors, pallor, anxiety, hyperglycemia
✅ Diagnosis:
- ↑ Plasma/urine metanephrines
- Imaging: CT/MRI, MIBG scan
✅ Preoperative Management:
- Alpha-blocker (phenoxybenzamine) first
- Then beta-blocker (after alpha-blockade)
- Ensure volume expansion
✅ Definitive Treatment:
- Laparoscopic or open adrenalectomy
✅ Postoperative Monitoring:
- For hypotension, hypoglycemia, and adrenal insufficiency
✅ Complications:
- Hypertensive crisis, stroke, arrhythmia, cardiomyopathy, and post-op adrenal shock
✅ Nursing Role:
- Monitor vitals closely
- Prevent triggers
- Prepare patient for surgery and lifelong follow-up
- Educate on medication adherence, genetic counseling, and emergency awareness
🧪 HYPERALDOSTERONISM
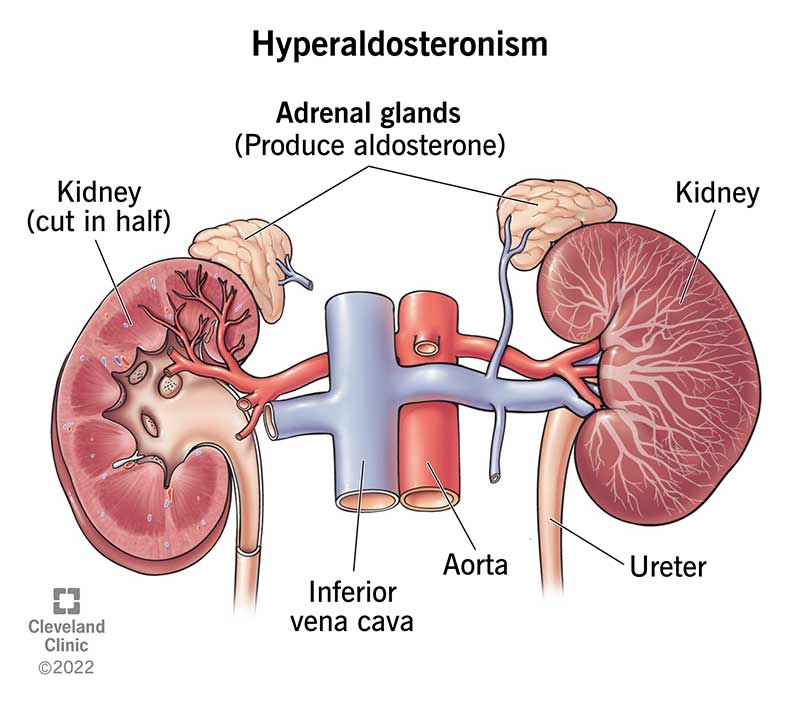
📌 Definition:
Hyperaldosteronism is a condition characterized by the excessive secretion of aldosterone, a mineralocorticoid hormone produced by the adrenal cortex, leading to sodium retention, potassium excretion, and hypertension.
🔄 Aldosterone acts on the kidneys to conserve sodium and excrete potassium. Its overproduction causes volume overload, hypokalemia, and high blood pressure.
🧬 Causes:
| 🔍 Cause Category | Examples |
|---|---|
| Primary (Conn’s Syndrome) | – Aldosterone-producing adrenal adenoma |
- Bilateral adrenal hyperplasia
- Rare: adrenal carcinoma | | Secondary Hyperaldosteronism | – Renal artery stenosis
- Congestive heart failure
- Liver cirrhosis
- Nephrotic syndrome
(All lead to increased renin → ↑ aldosterone) | | Genetic/Familial | Familial hyperaldosteronism types I–III (rare) |
🏷️ Types of Hyperaldosteronism:
| Type | Description |
|---|---|
| Primary Hyperaldosteronism | Autonomous aldosterone production independent of renin; most often due to adrenal adenoma or hyperplasia |
| Secondary Hyperaldosteronism | Occurs as a response to increased renin from decreased renal perfusion or fluid loss |
| Glucocorticoid-remediable aldosteronism (GRA) | A genetic form of hyperaldosteronism controlled by glucocorticoids |
🔬 Pathophysiology:
- Increased aldosterone secretion → stimulates kidneys to:
- Retain sodium and water → ↑ blood volume → hypertension
- Excrete potassium → hypokalemia
- Excrete hydrogen ions → metabolic alkalosis
- Resulting hormonal imbalance leads to:
- Volume overload → increased blood pressure
- Electrolyte disturbances → muscle weakness, arrhythmias
- Suppression of renin (in primary type)
🚨 Signs and Symptoms:
| System | Symptoms |
|---|---|
| Cardiovascular | – Hypertension (often resistant to treatment) |
- Headaches, flushing | | Neuromuscular | – Muscle cramps, weakness
- Fatigue
- Numbness or tingling (paresthesias) | | Renal/Metabolic | – Hypokalemia → polyuria, polydipsia
- Nocturia
- Metabolic alkalosis | | Cardiac arrhythmias | – Due to low potassium (e.g., premature beats, tachycardia) |
🧪 Diagnosis:
| Test | Findings |
|---|---|
| Plasma aldosterone concentration (PAC) | ↑ Elevated |
| Plasma renin activity (PRA) | ↓ Suppressed (in primary) |
| Aldosterone-renin ratio (ARR) | ↑ High ratio (>20:1) suggests primary hyperaldosteronism |
| Serum electrolytes | ↓ Potassium, ↑ Sodium, ↑ Bicarbonate |
| 24-hour urinary aldosterone | Confirms aldosterone excess |
| CT/MRI of adrenal glands | Detects adrenal adenoma or hyperplasia |
| Adrenal vein sampling (AVS) | Differentiates unilateral vs bilateral disease |
| Saline or fludrocortisone suppression test | Confirms autonomous aldosterone production |
💊 Medical Management:
| Treatment | Purpose |
|---|---|
| Mineralocorticoid receptor antagonists | |
| (e.g., Spironolactone, Eplerenone) | – Block aldosterone effects |
- Used in bilateral adrenal hyperplasia and unilateral cases not suitable for surgery | | Antihypertensives | – Control BP if not normalized with spironolactone | | Potassium supplements | – Correct hypokalemia | | Low-sodium diet | – Helps control blood pressure |
✂️ Surgical Management:
🎯 Definitive treatment for unilateral adrenal adenoma (Conn’s syndrome)
| Surgery | Description |
|---|---|
| Laparoscopic adrenalectomy | – Removal of the affected adrenal gland |
- Normalizes BP and potassium in most patients | | Bilateral hyperplasia | Surgery not preferred → medical management with spironolactone | | Post-op care | Monitor for hypotension, electrolyte shifts, and renal function changes |
🩺 Nursing Management:
| Focus Area | Nursing Action |
|---|---|
| Monitor BP and HR regularly | Detect hypertension and response to treatment |
| Assess for hypokalemia symptoms | Muscle cramps, fatigue, cardiac irregularities |
| Daily weight and I&O | Monitor fluid balance and signs of fluid overload |
| Administer medications as ordered | Spironolactone, potassium supplements, antihypertensives |
| Educate on low-sodium, high-potassium diet | Supports medical therapy |
| Pre- and post-op care (if surgery done) | BP control, infection prevention, hormone monitoring |
| Patient education | Medication adherence, need for regular follow-up, lifestyle changes |
⚠️ Complications:
- Resistant hypertension → risk of stroke, heart attack, heart failure
- Severe hypokalemia → muscle paralysis, life-threatening arrhythmias
- Metabolic alkalosis
- Renal damage due to long-standing hypertension
- Postoperative adrenal insufficiency (if bilateral adrenalectomy is done)
🧷 Key Points on Hyperaldosteronism
✅ Definition: Excess aldosterone → sodium retention, potassium loss, hypertension
✅ Primary cause: Adrenal adenoma (Conn’s Syndrome), bilateral adrenal hyperplasia
✅ Secondary cause: Due to ↑ renin from conditions like renal artery stenosis, CHF
✅ Classic features:
- Hypertension + Hypokalemia + Metabolic alkalosis
✅ Diagnosis:
- ↑ Aldosterone
- ↓ Renin (in primary)
- CT scan, AVS, suppression tests
✅ Treatment:
- Spironolactone or eplerenone
- Adrenalectomy (if unilateral tumor)
- Lifestyle: low-salt diet, potassium-rich food
✅ Nursing care:
- Monitor BP, electrolytes
- Educate on medication, diet, signs of crisis
- Ensure adherence to follow-up and therapy
🧬 ADRENAL INSUFFICIENCY
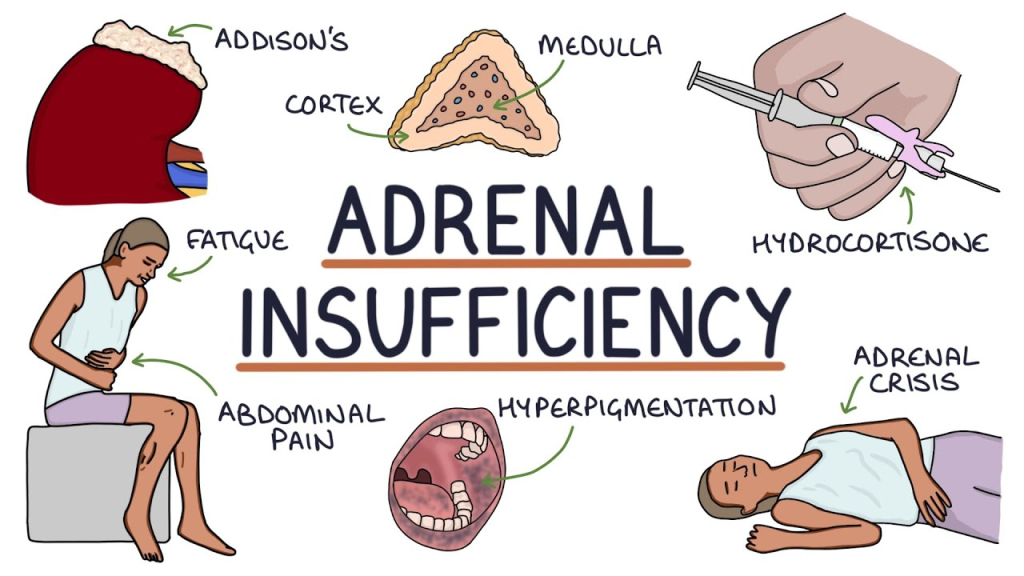
📌 Definition:
Adrenal insufficiency is a condition in which the adrenal glands fail to produce adequate amounts of hormones, particularly cortisol (and sometimes aldosterone). Cortisol is essential for stress response, metabolism, immune function, and blood pressure regulation.
🛑 In severe cases, it can lead to an Addisonian crisis—a life-threatening emergency.
🧬 Causes:
| 🔍 Category | Examples |
|---|---|
| Primary Adrenal Insufficiency (Addison’s Disease) | – Autoimmune adrenalitis |
- Tuberculosis
- Adrenal hemorrhage (Waterhouse-Friderichsen syndrome)
- Adrenalectomy
- Metastatic cancer to adrenal glands
- Genetic enzyme deficiencies (e.g., CAH) | | Secondary Adrenal Insufficiency | – Pituitary tumors or surgery
- Traumatic brain injury
- Sheehan’s syndrome
- Long-term corticosteroid therapy withdrawal | | Tertiary Adrenal Insufficiency | – Hypothalamic damage
- Suppressed CRH due to prolonged steroid use
- Brain tumors affecting hypothalamus |
🏷️ Types of Adrenal Insufficiency:
| Type | Description |
|---|---|
| Primary (Addison’s Disease) | Destruction of the adrenal cortex → ↓ cortisol + ↓ aldosterone |
| Secondary | Pituitary failure → ↓ ACTH → ↓ cortisol (aldosterone typically preserved) |
| Tertiary | Hypothalamic failure or abrupt steroid withdrawal → ↓ CRH → ↓ ACTH → ↓ cortisol |
🔬 Pathophysiology:
- ↓ Cortisol production → impaired glucose regulation, immune response, stress tolerance
- ↓ Aldosterone (in primary) → sodium loss, potassium retention, hypovolemia, hypotension
- Feedback disruption → ↑ ACTH in primary, ↓ ACTH in secondary
- ACTH ↑ → stimulates melanocytes → hyperpigmentation (only in primary)
- Result: fatigue, hypotension, electrolyte imbalance, weight loss, and crisis during stress
🚨 Signs and Symptoms:
| System | Symptoms |
|---|---|
| General | Weakness, fatigue, weight loss |
| Skin | Hyperpigmentation (primary only), especially in skin folds, gums |
| Cardiovascular | Hypotension, orthostatic hypotension |
| GI | Nausea, vomiting, diarrhea, abdominal pain |
| Neuropsychiatric | Irritability, depression, confusion |
| Renal/Electrolyte | Hyponatremia, hyperkalemia (primary), dehydration |
| Others | Craving for salty foods, hypoglycemia (especially in children) |
🧪 Diagnosis:
| Test | Findings |
|---|---|
| Morning serum cortisol | ↓ Low (especially at 8 a.m.) |
| Plasma ACTH | ↑ High in primary, ↓ or normal in secondary |
| ACTH stimulation test (Cosyntropin test) | No cortisol rise in primary; delayed/partial in secondary |
| Serum electrolytes | ↓ Sodium, ↑ Potassium, ↓ Glucose |
| Renin & aldosterone | ↑ Renin, ↓ Aldosterone (in primary) |
| Imaging | CT/MRI of adrenal glands or pituitary (to identify cause) |
💊 Medical Management:
| Medication | Purpose |
|---|---|
| Hydrocortisone (oral or IV) | First-line glucocorticoid replacement |
| Fludrocortisone | Mineralocorticoid replacement (only in primary adrenal insufficiency) |
| Dexamethasone/Prednisolone | Alternatives for long-term steroid therapy |
| IV fluids + dextrose | In adrenal crisis to treat dehydration, hypotension, and hypoglycemia |
| Electrolyte correction | Manage hyperkalemia, hyponatremia |
🛠️ Surgical Management:
- Not commonly used, but may be necessary in:
- Adrenal tumors or hemorrhage
- Pituitary tumors causing secondary adrenal insufficiency
- Bilateral adrenalectomy for certain endocrine conditions (e.g., Cushing’s)
🩺 Nursing Management:
| Focus Area | Nursing Interventions |
|---|---|
| Vital signs monitoring | Watch for hypotension, bradycardia, dehydration |
| Monitor glucose and electrolytes | Correct hypoglycemia, hyponatremia, hyperkalemia |
| Administer medications as prescribed | Hydrocortisone, fludrocortisone |
| Stress dosing education | Teach patient to increase steroid dose during illness or surgery |
| Fluid balance | Monitor I&O, maintain hydration |
| Skin care | For hyperpigmentation, dryness |
| Educate patient and family | Disease process, signs of crisis, medication adherence, emergency steroid kit use |
| Medical alert bracelet | Encourage wearing for emergency identification |
⚠️ Complications:
- Addisonian Crisis: acute adrenal failure → shock, coma, death
- Electrolyte imbalances: hyperkalemia, hyponatremia
- Severe hypotension: resistant to fluids alone
- Hypoglycemia
- Adrenal crisis during surgery/infection if not stress-dosed
🧷 Key Points Summary:
✅ Adrenal insufficiency = ↓ cortisol (± aldosterone)
✅ Primary = adrenal gland problem → ↑ ACTH, hyperpigmentation
✅ Secondary = pituitary problem → ↓ ACTH, no hyperpigmentation
✅ Symptoms: fatigue, hypotension, salt craving, GI issues, electrolyte imbalances
✅ Diagnosed via ACTH stimulation test, cortisol & ACTH levels
✅ Treated with glucocorticoids, mineralocorticoids (if primary)
✅ Crisis = emergency → needs IV hydrocortisone + fluids
✅ Nurses monitor vitals, electrolytes, and teach lifelong steroid management
🧬 ADRENAL TUMORS
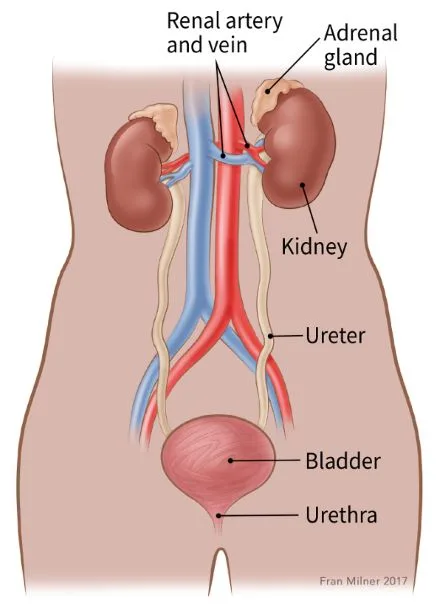
📌 Definition:
Adrenal tumors are abnormal growths that develop in the adrenal glands, which are located above each kidney and consist of two parts:
- Adrenal cortex (outer part): produces cortisol, aldosterone, and androgens
- Adrenal medulla (inner part): produces catecholamines (epinephrine and norepinephrine)
Tumors may be benign or malignant, functional (hormone-secreting) or non-functional.
🧬 Causes:
| Cause | Description |
|---|---|
| Unknown (sporadic) | Most adrenal tumors are spontaneous and idiopathic |
| Genetic syndromes | – Multiple Endocrine Neoplasia (MEN) type 2 |
- Li-Fraumeni syndrome
- Beckwith-Wiedemann syndrome
- Von Hippel-Lindau (VHL) disease
- Neurofibromatosis type 1 (NF1) | | Adrenal hyperplasia | May develop into nodules or tumors | | Metastatic spread | Cancer from lung, breast, or kidney may spread to adrenal glands |
🏷️ Types of Adrenal Tumors:
| Tumor Type | Origin | Hormone Status | Common Names |
|---|---|---|---|
| Adenoma | Cortex | Functional or non-functional | Benign; may cause Cushing’s or Conn’s syndrome |
| Adrenocortical carcinoma | Cortex | Often functional | Rare, aggressive |
| Pheochromocytoma | Medulla | Functional | Secretes catecholamines |
| Neuroblastoma | Medulla | Often non-functional | Common in children |
| Myelolipoma | Cortex | Non-functional | Benign, fatty tumor |
| Metastatic adrenal tumors | Secondary | Usually non-functional | From breast, lung, GI tract |
🔬 Pathophysiology:
- Uncontrolled cellular growth in the adrenal cortex or medulla forms a mass.
- If functional, the tumor secretes excess hormones:
- Cortisol → Cushing’s syndrome
- Aldosterone → Hyperaldosteronism (Conn’s syndrome)
- Androgens/estrogens → Virilization/feminization
- Catecholamines → Pheochromocytoma symptoms
- These hormones disrupt normal metabolic, cardiovascular, and immune regulation.
- Non-functional tumors may grow without symptoms and are often found incidentally.
🚨 Signs and Symptoms:
Symptoms depend on the hormone secreted or tumor size
✅ If Functional:
| Hormone | Symptoms |
|---|---|
| Cortisol (Cushing’s) | Weight gain, moon face, purple striae, muscle weakness, hyperglycemia |
| Aldosterone (Conn’s) | Hypertension, hypokalemia, muscle cramps |
| Androgens/Estrogens | Hirsutism, acne, deep voice in females; gynecomastia in males |
| Catecholamines (Pheochromocytoma) | Headaches, sweating, palpitations, paroxysmal hypertension |
✅ If Non-functional or Large:
- Flank or abdominal pain
- Palpable abdominal mass
- Pressure symptoms on nearby organs
- Often asymptomatic
🧪 Diagnosis:
| Test | Purpose |
|---|---|
| Hormonal blood tests | Cortisol, ACTH, aldosterone, renin, androgens, catecholamines |
| 24-hour urine tests | Metanephrines, cortisol, VMA |
| Imaging | CT scan or MRI of the abdomen to locate and size tumor |
| PET scan / MIBG scan | To evaluate malignancy or locate pheochromocytoma |
| Adrenal vein sampling | Helps localize source in bilateral disease |
| Biopsy | Rarely done—only if metastatic disease is suspected and hormone secretion is ruled out |
💊 Medical Management:
- Preoperative stabilization (especially for functional tumors like pheochromocytoma)
- Alpha-blockers: phenoxybenzamine
- Beta-blockers: propranolol (after alpha-blockade)
- Spironolactone: for hyperaldosteronism
- Cortisol suppression: Ketoconazole, metyrapone (for cortisol-producing tumors)
- Electrolyte correction: Potassium, glucose, and BP control
- Hormone replacement post-surgery (especially if bilateral adrenalectomy)
✂️ Surgical Management:
🎯 Surgery is the definitive treatment for most adrenal tumors
| Type | Details |
|---|---|
| Laparoscopic adrenalectomy | Preferred for benign, localized, and small tumors |
| Open adrenalectomy | Indicated for large, invasive, or malignant tumors |
| Bilateral adrenalectomy | For bilateral disease or Cushing’s refractory to medical treatment |
| Post-op care | Hormonal monitoring, steroids if both glands removed |
🩺 Nursing Management:
| Focus Area | Interventions |
|---|---|
| Pre-op care | BP control, monitor electrolytes, initiate alpha-blockade if pheochromocytoma |
| Post-op monitoring | Vitals, fluid balance, electrolyte levels, glucose, signs of adrenal insufficiency |
| Medication administration | Steroids, antihypertensives, hormone inhibitors |
| Pain & wound care | Standard post-surgical management |
| Psychological support | Body image, anxiety, chronic illness adjustment |
| Patient education | Lifelong hormone therapy if adrenalectomy, follow-up, emergency steroid use |
⚠️ Complications:
- Adrenal crisis post-surgery (especially bilateral adrenalectomy)
- Hormonal imbalances → Cushing’s, Conn’s, virilization
- Hypertensive crisis (pheochromocytoma)
- Recurrence or metastasis (especially in adrenocortical carcinoma)
- Post-op complications: bleeding, infection, DVT
- Psychosocial impact: from body changes, long-term medication use
🧷 Key Points Summary:
✅ Adrenal tumors can arise from cortex or medulla, be benign/malignant, and functional/non-functional
✅ Common types: adenoma, pheochromocytoma, adrenocortical carcinoma
✅ Symptoms vary by hormone secreted—cortisol, aldosterone, androgens, catecholamines
✅ Diagnosed via hormonal assays + imaging
✅ Treatment includes surgery, hormone control, and lifelong follow-up
✅ Nurses play a key role in preparing, monitoring, educating, and preventing crises
🧠 DISORDERS OF THE PITUITARY GLAND
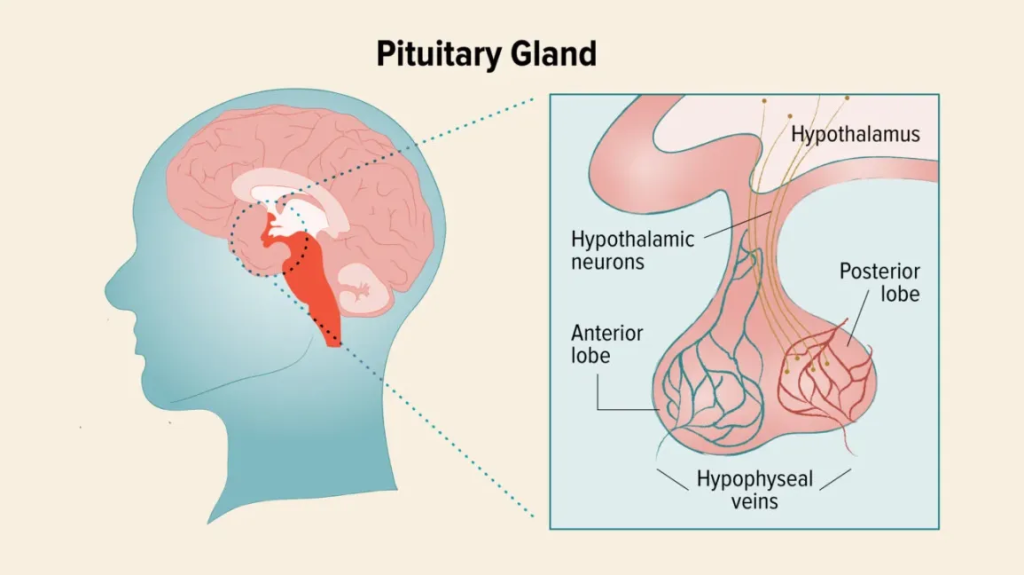
📌 Overview of the Pituitary Gland:
The pituitary gland, also known as the “master gland,” is a small pea-sized endocrine gland located at the base of the brain in the sella turcica. It is divided into:
- Anterior lobe (adenohypophysis) – secretes GH, TSH, ACTH, FSH, LH, and prolactin
- Posterior lobe (neurohypophysis) – stores and releases ADH and oxytocin
Disorders can result from hyposecretion or hypersecretion of hormones or from tumors and trauma affecting its function.
🏷️ Types of Pituitary Disorders:
| Category | Common Disorders |
|---|---|
| Anterior Pituitary Disorders | – Hyperpituitarism (e.g., acromegaly, gigantism, Cushing’s disease, prolactinoma) |
- Hypopituitarism (partial or complete)
- Pituitary tumors | | Posterior Pituitary Disorders | – Diabetes Insipidus (DI)
- Syndrome of Inappropriate Antidiuretic Hormone (SIADH) |
🧬 Causes:
- Pituitary adenomas (benign tumors; most common)
- Craniopharyngioma (in children)
- Trauma or surgery to the brain/pituitary
- Radiation therapy
- Infections (e.g., meningitis, TB)
- Autoimmune hypophysitis
- Congenital defects
- Sheehan’s syndrome (postpartum pituitary infarction)
🔬 Common Pituitary Disorders in Detail:
1️⃣ Hyperpituitarism (Hormone Overproduction)
🧪 Most often due to pituitary adenomas
Depending on the hormone involved:
- Acromegaly/Gigantism: GH excess → enlarged hands/feet, coarse facial features, tall stature (gigantism if before puberty)
- Cushing’s Disease: ACTH excess → cortisol overproduction (moon face, striae, obesity)
- Prolactinoma: Excess prolactin → galactorrhea, infertility, amenorrhea
2️⃣ Hypopituitarism (Hormone Deficiency)
Can be partial (1–2 hormones) or panhypopituitarism (all hormones)
- ↓ GH → growth failure in children, fatigue in adults
- ↓ TSH → secondary hypothyroidism
- ↓ ACTH → secondary adrenal insufficiency
- ↓ FSH/LH → infertility, amenorrhea, loss of libido
- ↓ Prolactin → failure to lactate post-delivery
3️⃣ Pituitary Tumors
- Most are benign adenomas
- Can be functional (hormone-secreting) or non-functional
- May cause mass effect: headache, vision changes (optic chiasm compression → bitemporal hemianopia), increased ICP
4️⃣ Diabetes Insipidus (Posterior Pituitary)
- ↓ ADH → polyuria, polydipsia, dilute urine
- Caused by head injury, tumors, or surgery
5️⃣ SIADH (Syndrome of Inappropriate ADH Secretion)
- ↑ ADH → water retention → hyponatremia, confusion, seizures
- Often associated with brain injury, tumors, or lung carcinoma
🧪 Diagnosis of Pituitary Disorders:
- Hormonal blood levels (GH, TSH, ACTH, prolactin, cortisol, FSH, LH)
- 24-hour urine for cortisol (in Cushing’s)
- Water deprivation test (for DI)
- MRI/CT of the pituitary gland
- Visual field testing (if optic chiasm involvement)
- Stimulation/suppression tests (e.g., insulin tolerance test)
💊 Medical Management:
- Hormone replacement therapy: Hydrocortisone, levothyroxine, sex hormones, desmopressin (for DI)
- Dopamine agonists (e.g., cabergoline, bromocriptine) for prolactinomas
- Somatostatin analogs (e.g., octreotide) for GH suppression in acromegaly
- Fluid restriction, diuretics (e.g., furosemide) in SIADH
- Electrolyte correction
✂️ Surgical Management:
- Transsphenoidal surgery: Preferred approach for pituitary adenomas
- Craniotomy: For large or invasive tumors
- Post-op: monitor for CSF leak, diabetes insipidus, pituitary hormone deficits
🩺 Nursing Management:
| Area | Nursing Actions |
|---|---|
| Monitoring | Vital signs, neuro status, intake/output, signs of hormone imbalance |
| Post-op care | Prevent sneezing, coughing, straining; monitor for CSF leak (clear nasal discharge), DI signs |
| Hormonal therapy | Administer and monitor replacement hormones |
| Patient education | Lifelong hormone therapy, signs of crisis, when to seek help |
| Fluid balance | Especially in SIADH and DI |
| Emotional support | Coping with chronic illness, body image issues (e.g., acromegaly, Cushing’s) |
⚠️ Complications:
- Adrenal crisis, thyroid crisis
- Pituitary apoplexy (hemorrhage/infarction of the gland)
- Infertility, visual disturbances, depression
- Water intoxication or dehydration (SIADH or DI)
- Hypopituitarism after surgery or radiation
🧷 Key Points Summary:
✅ Pituitary gland controls multiple endocrine functions
✅ Disorders can be due to excess or deficiency of hormone secretion
✅ Common conditions: Cushing’s disease, acromegaly, prolactinoma, DI, SIADH
✅ Diagnosed via hormone levels, MRI, stimulation/suppression tests
✅ Treated with medications, surgery, and lifelong hormone replacement
✅ Nurses play a crucial role in monitoring, post-op care, and patient education
🧠 ACROMEGALY
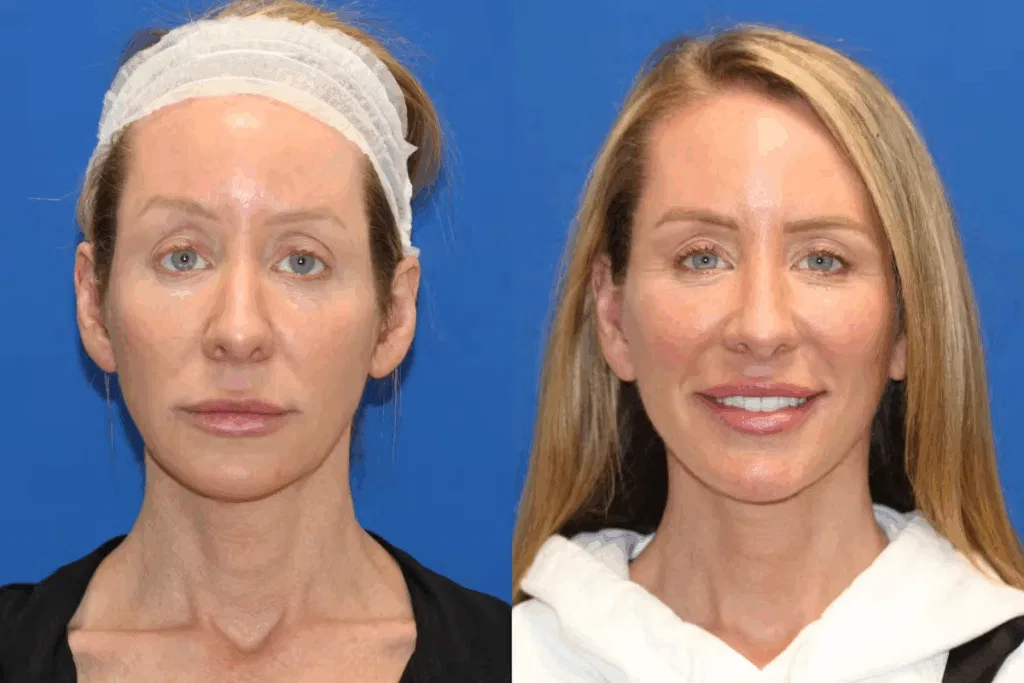
📌 Definition:
Acromegaly is a chronic hormonal disorder characterized by excessive secretion of growth hormone (GH) in adults (after the closure of epiphyseal growth plates), usually due to a pituitary adenoma. This results in progressive enlargement of bones and soft tissues, especially in the hands, feet, and face.
🧒 In children, GH excess before epiphyseal closure causes gigantism, not acromegaly.
🧬 Causes of Acromegaly:
| Category | Example |
|---|---|
| Pituitary causes (most common) | – GH-secreting pituitary adenoma (>95% cases) |
- Pituitary hyperplasia | | Non-pituitary (ectopic) GH/ GHRH secretion | – GH or GHRH-secreting tumors (e.g., lung or pancreatic tumors)
- Carcinoid tumors | | Genetic or familial syndromes | – MEN type 1
- McCune-Albright syndrome (rare) |
🏷️ Types of Acromegaly:
| Type | Description |
|---|---|
| Pituitary acromegaly | Due to benign adenoma of pituitary gland (most common form) |
| Ectopic acromegaly | Due to non-pituitary tumors secreting GH or GHRH (rare) |
| Familial acromegaly | Genetic syndromes with pituitary tumors (MEN1, AIP mutation) |
| Acromegaly + gigantism | Occurs if GH excess begins before and continues after puberty (rare overlap condition) |
🧬 Pathophysiology:
- A pituitary adenoma (GH-secreting tumor) or ectopic tumor secretes excess growth hormone (GH) in adults.
- GH stimulates the liver and other tissues to produce insulin-like growth factor 1 (IGF-1), which is responsible for most of the growth-promoting effects.
- Excess GH and IGF-1 lead to:
- Overgrowth of bones and soft tissues (especially hands, feet, face)
- Organomegaly (heart, liver, kidneys)
- Metabolic changes like insulin resistance → diabetes
- Increased cartilage and bone thickening, especially in skull, jaw, and vertebrae
- Compression effects from the pituitary tumor may also affect nearby structures:
- Optic chiasm → visual field defects
- Normal pituitary → hypopituitarism
🚨 Signs and Symptoms of Acromegaly:
Slow onset; symptoms develop over years, often missed early.
🦴 Skeletal & Soft Tissue Changes:
- Enlargement of hands and feet (rings/shoes don’t fit)
- Coarsened facial features: protruding jaw (prognathism), enlarged nose, lips, ears
- Widened fingers, thick skin, oily skin
- Increased hat, shoe, glove size
💓 Cardiovascular:
- Hypertension
- Cardiomegaly, left ventricular hypertrophy
- Heart failure in late stages
🧠 Neurological:
- Headaches
- Visual field defects (especially bitemporal hemianopia due to optic chiasm compression)
- Peripheral neuropathy (carpal tunnel syndrome)
🍬 Metabolic:
- Insulin resistance → diabetes mellitus
- Hyperlipidemia
🧬 Reproductive:
- Menstrual irregularities, infertility in women
- Decreased libido, erectile dysfunction in men
- Galactorrhea (due to concurrent prolactin elevation)
😴 Other Symptoms:
- Sleep apnea
- Fatigue, joint pain, voice deepening
🧪 Diagnosis of Acromegaly:
✅ Hormonal Tests:
| Test | Interpretation |
|---|---|
| Serum IGF-1 level | ↑ Elevated (most reliable screening test) |
| GH suppression test (Oral glucose tolerance test – OGTT) | GH fails to suppress after glucose load = positive for acromegaly |
| Random GH level | May be elevated, but variable; less reliable alone |
✅ Imaging:
| Imaging | Purpose |
|---|---|
| MRI of pituitary gland | Detects pituitary adenoma (most common cause) |
| CT scan | Used if MRI is contraindicated or to look for ectopic tumors |
✅ Visual and Other Assessments:
- Visual field testing: To detect optic nerve compression (bitemporal hemianopia)
- Blood glucose and HbA1c: Evaluate for diabetes
- Echocardiography: Detect cardiomyopathy
- Sleep studies: Assess for obstructive sleep apnea
💊 I. MEDICAL MANAGEMENT
🎯 Goals:
- Normalize GH and IGF-1 levels
- Control tumor growth
- Relieve symptoms
- Prevent complications like diabetes, heart disease, and arthritis
✅ 1. Somatostatin Analogs (First-line medical therapy)
| Drug | Action |
|---|---|
| Octreotide (Sandostatin) | |
| Lanreotide (Somatuline) | – Mimic natural somatostatin |
- Suppress GH secretion from pituitary tumor
- Reduce IGF-1 levels
- May shrink tumor size in some patients |
✅ 2. GH Receptor Antagonist
| Drug | Action |
|---|---|
| Pegvisomant | – Blocks GH receptors in the liver |
- Reduces IGF-1 levels
- Does not reduce tumor size, used if somatostatin analogs fail |
✅ 3. Dopamine Agonists
| Drug | Action |
|---|---|
| Cabergoline, Bromocriptine | – Inhibit GH secretion |
- Less effective than somatostatin analogs
- More effective if prolactin is also elevated |
✅ 4. Supportive Management
- Antihypertensives – for high blood pressure
- Oral hypoglycemics or insulin – for diabetes
- Sleep therapy – for obstructive sleep apnea
- Pain relief and physical therapy – for joint pain
🛠️ II. SURGICAL MANAGEMENT
🎯 Transsphenoidal surgery is the treatment of choice for GH-secreting pituitary tumors.
🔪 1. Transsphenoidal Adenomectomy
- Minimally invasive surgery through the nose and sphenoid sinus
- Removes pituitary adenoma causing GH excess
- Success rate depends on tumor size and surgeon skill
Benefits:
- Immediate drop in GH/IGF-1 levels (if successful)
- Preserves normal pituitary function
🛑 Surgical Risks:
- Bleeding
- CSF leak
- Diabetes insipidus (transient or permanent)
- Damage to normal pituitary tissue → hypopituitarism
🔄 2. Repeat Surgery / Debulking
- For residual or recurrent tumors not completely removed in the first surgery
💡 3. Stereotactic Radiotherapy / Gamma Knife
- Used when:
- Surgery is not possible or fails
- Patient is not a surgical candidate
- Tumor is invasive
Downsides:
- Slow to act (months to years)
- May cause hypopituitarism
🩺 NURSING MANAGEMENT OF ACROMEGALY
🎯 Nursing Goals:
- Monitor for and manage hormonal imbalances
- Support recovery post surgery or medical therapy
- Prevent complications such as diabetes, hypertension, visual changes
- Provide emotional support and education for lifelong care
🗂️ I. Nursing Assessment
| Area | Key Assessment |
|---|---|
| Vital signs | Monitor for hypertension, tachycardia |
| Neuro checks | Monitor for headache, visual disturbances, signs of increased ICP |
| Respiratory status | Observe for snoring, sleep apnea symptoms |
| Blood glucose levels | Monitor for hyperglycemia |
| Physical changes | Assess for enlargement of extremities, joint pain, fatigue |
| Psychosocial | Screen for depression, self-esteem issues due to altered body image |
📝 II. Common Nursing Diagnoses
- Body image disturbance related to facial and physical changes
- Chronic pain related to joint and skeletal overgrowth
- Impaired vision related to optic chiasm compression
- Imbalanced nutrition: more than body requirements
- Knowledge deficit related to disease, lifelong treatment
- Risk for injury due to joint instability or visual field loss
🧾 III. Nursing Interventions
🔹 A. Preoperative Care (If Surgery Planned)
| Intervention | Purpose |
|---|---|
| Educate about transsphenoidal surgery | Reduce fear and promote cooperation |
| Explain expected outcomes and complications | Informed consent and realistic expectations |
| Maintain fluid balance and nutrition | Prepare for surgery and promote healing |
| Encourage psychological support | Reduce anxiety, support self-image issues |
🔹 B. Postoperative Care (Transsphenoidal Surgery)
| Intervention | Purpose |
|---|---|
| Monitor for CSF leak (clear nasal discharge) | Prevent meningitis |
| Avoid sneezing, coughing, straining | Prevent increased ICP or CSF leak |
| Neurological monitoring | Detect signs of brain involvement or complications |
| Assess for DI (excessive urination, thirst) | May occur due to pituitary manipulation |
| Administer prescribed hormone replacement therapy if needed | Treat postoperative hypopituitarism |
🔹 C. Long-Term & Supportive Care
| Intervention | Purpose |
|---|---|
| Monitor blood glucose and BP regularly | Control metabolic complications |
| Teach medication regimen (e.g., octreotide, pegvisomant) | Ensure adherence and reduce hormone levels |
| Encourage regular follow-up visits | Monitor IGF-1 levels, MRI scans |
| Provide referrals: endocrinologist, ophthalmologist, counselor | Holistic, multidisciplinary support |
| Support coping strategies | Address emotional effects of disfigurement and chronic illness |
📊 IV. Evaluation Criteria (Expected Outcomes):
- Patient maintains stable hormone levels (GH, IGF-1)
- Verbalizes understanding of disease and treatment
- Demonstrates adherence to medications and follow-up
- Reports improved coping and reduced stress
- Free from postoperative or treatment-related complications
⚠️ COMPLICATIONS OF ACROMEGALY
Uncontrolled or late-diagnosed acromegaly can lead to serious systemic complications, many of which are irreversible without early intervention.
🧠 1. Neurological Complications:
- Headaches due to tumor pressure
- Bitemporal hemianopia from optic chiasm compression
- Carpal tunnel syndrome from nerve compression
- Peripheral neuropathy
❤️ 2. Cardiovascular Complications:
- Hypertension (very common)
- Cardiomyopathy → heart failure
- Arrhythmias
- Increased risk of stroke & sudden cardiac death
🍬 3. Metabolic Complications:
- Insulin resistance → Type 2 Diabetes Mellitus
- Hyperlipidemia
- Weight gain/obesity
🦴 4. Musculoskeletal Issues:
- Joint pain, arthritis
- Kyphosis, enlarged hands and feet
- Osteoarthritis due to cartilage overgrowth
👃 5. Respiratory Complications:
- Obstructive sleep apnea due to soft tissue overgrowth in airway
- Voice deepening and snoring
🧬 6. Reproductive & Endocrine Disorders:
- Menstrual irregularities, infertility
- Erectile dysfunction, decreased libido
- Galactorrhea (if prolactin is elevated)
🧪 7. Post-Treatment Complications:
- Hypopituitarism (especially post-surgery or radiotherapy)
- CSF leak or infection after transsphenoidal surgery
- Adrenal or thyroid insufficiency if entire pituitary function is impaired
🧷 KEY POINTS ON ACROMEGALY
✅ Definition: A hormonal disorder in adults due to excess GH secretion, usually from a pituitary adenoma
✅ Key hormones involved: Growth hormone (GH) and IGF-1
✅ Classic signs:
- Enlarged hands, feet, facial features
- Joint pain, coarsened skin, voice deepening
- Hypertension, diabetes, sleep apnea
✅ Diagnosis:
- ↑ IGF-1 levels, OGTT GH suppression test
- MRI of pituitary gland
✅ Treatment:
- Transsphenoidal surgery (first-line)
- Somatostatin analogs, GH receptor antagonists
- Radiotherapy if tumor is unresectable
✅ Nursing role:
- Monitor for visual changes, blood sugar, BP
- Post-op care to prevent CSF leak, DI
- Provide psychosocial support and promote medication adherence
✅ Complications:
- Diabetes, heart disease, neuropathy, sleep apnea, infertility, and hypopituitarism
📏 GIGANTISM
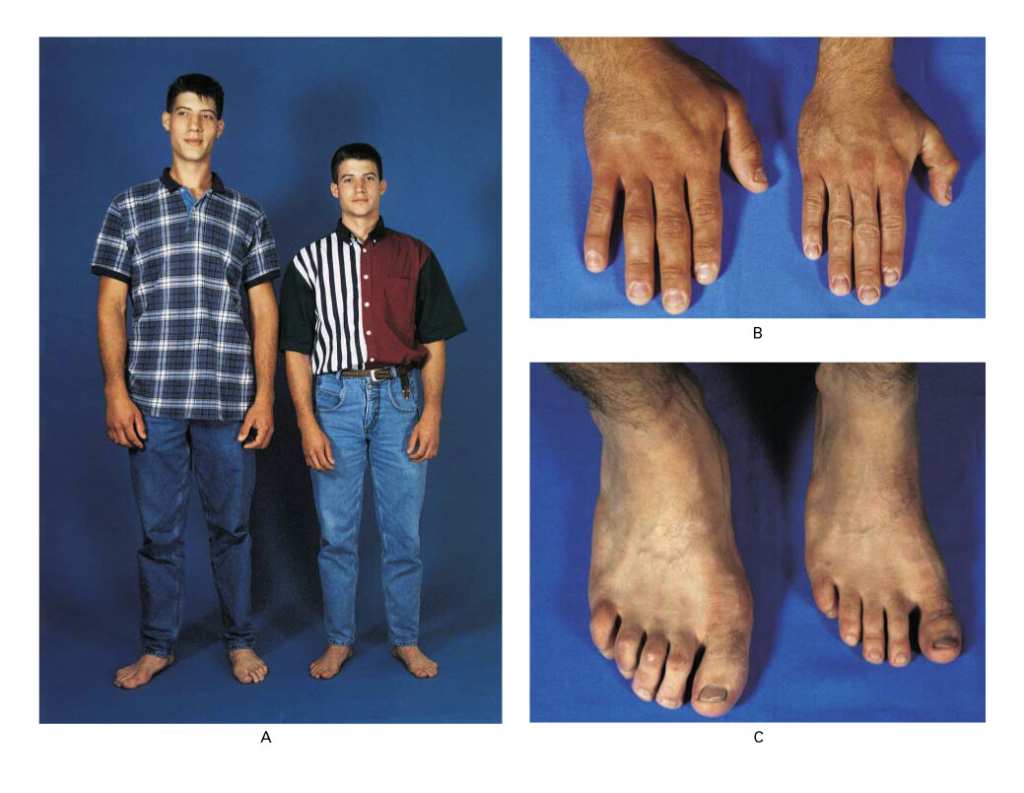
📌 Definition:
Gigantism is a rare endocrine disorder characterized by excessive secretion of growth hormone (GH) before the epiphyseal growth plates close (i.e., during childhood or adolescence), resulting in abnormally increased linear growth and tall stature.
🧠 It is caused by the same mechanisms as acromegaly, but it occurs before puberty, leading to height >97th percentile for age and sex.
🧬 Causes of Gigantism:
| Category | Examples |
|---|---|
| Pituitary causes | – GH-secreting pituitary adenoma (most common) |
- Pituitary hyperplasia
- Craniopharyngioma | |
- Hypothalamic causes | – Excess GHRH secretion from hypothalamic tumors | |
- Genetic syndromes | – McCune-Albright syndrome, MEN type 1, Carney complex | |
- Other rare causes | – Ectopic GH or GHRH-secreting tumors (e.g., pancreatic, bronchial tumors)
- Gigantism secondary to neurofibromatosis or genetic overgrowth syndromes (e.g., Sotos syndrome) |
🏷️ Types of Gigantism:
| Type | Description |
|---|---|
| Pituitary Gigantism | Most common type due to GH-secreting pituitary adenoma in children |
| Hypothalamic Gigantism | Due to excess GHRH secretion, stimulating pituitary GH production |
| Ectopic Gigantism | Caused by non-pituitary tumors that secrete GH or GHRH (very rare) |
| Familial or Genetic Gigantism | Occurs in hereditary syndromes like MEN-1, often with multiple endocrine tumors |
| Cerebral Gigantism (Sotos Syndrome) | Genetic overgrowth syndrome not caused by GH excess but presents with tall stature and advanced bone age |
🧬 Pathophysiology:
- Excess secretion of growth hormone (GH) occurs before epiphyseal (growth) plate closure, typically during childhood or adolescence.
- GH stimulates the liver and peripheral tissues to produce insulin-like growth factor 1 (IGF-1), the main mediator of growth.
- IGF-1 promotes:
- Increased chondrocyte activity in growth plates → excessive linear bone growth
- Growth of soft tissues, organs, and metabolism
- The pituitary tumor or other sources (e.g., ectopic GHRH-producing tumors) often cause:
- Progressive enlargement of skeletal and soft tissues
- Organomegaly
- Metabolic dysregulation (e.g., insulin resistance, hyperglycemia)
⚠️ If GH excess continues into adulthood (after plate closure), it leads to acromegaly instead of gigantism.
🚨 Signs and Symptoms of Gigantism:
Symptoms typically begin in early childhood or adolescence, progressing gradually.
📏 Skeletal and Growth Abnormalities:
- Abnormally tall stature (height >97th percentile for age and sex)
- Rapid linear growth far above age expectations
- Long arms and legs, large hands and feet
- Enlarged jaw (prognathism), facial bone overgrowth
- Frontal bossing, increased head circumference
💪 Musculoskeletal Symptoms:
- Joint pain, arthritis due to skeletal overgrowth
- Muscle weakness despite size
- Kyphosis or scoliosis
❤️ Cardiovascular Symptoms:
- Hypertension
- Cardiomegaly, risk of heart failure
🍬 Metabolic Symptoms:
- Hyperglycemia or insulin resistance
- Increased appetite, weight gain despite tall stature
🧠 Neurological Symptoms:
- Headache
- Visual field defects (especially bitemporal hemianopia from optic chiasm compression by pituitary tumor)
- Delayed puberty or menstrual irregularities
👁️ Other Symptoms:
- Excessive sweating, thick skin
- Voice deepening in males
- Emotional/behavioral changes in adolescents
🧪 Diagnosis of Gigantism:
✅ 1. Hormonal Evaluation:
| Test | Expected Findings |
|---|---|
| Serum IGF-1 | ↑ Elevated (most reliable marker) |
| Random GH level | May be ↑ but fluctuates—less reliable alone |
| Oral glucose tolerance test (OGTT) | GH fails to suppress after glucose → diagnostic of GH excess |
| Serum Prolactin | May be elevated if tumor also secretes prolactin |
✅ 2. Imaging Studies:
| Test | Purpose |
|---|---|
| MRI of brain (pituitary focus) | Detects pituitary adenoma or hyperplasia |
| CT/MRI of other regions | Rule out ectopic GH/GHRH-secreting tumors (e.g., lung, pancreas) |
✅ 3. Other Investigations:
- X-rays: show enlarged bones, open epiphyses
- Visual field testing: to detect optic chiasm involvement
- Bone age studies: confirm advanced bone development
- Genetic testing: if a familial or syndromic cause is suspected (e.g., MEN1, McCune-Albright)
💊 I. MEDICAL MANAGEMENT
🎯 Objectives:
- Suppress or block excess growth hormone (GH) secretion
- Reduce IGF-1 levels
- Shrink the tumor if possible
- Prevent or manage metabolic and structural complications
✅ 1. Somatostatin Analogues (First-Line Medical Therapy)
| Drug | Action |
|---|---|
| Octreotide (Sandostatin) | |
| Lanreotide | – Mimic somatostatin, a natural GH inhibitor |
- Suppress GH secretion
- Lower IGF-1 levels
- May help shrink pituitary adenoma in some cases |
✅ 2. GH Receptor Antagonist
| Drug | Action |
|---|---|
| Pegvisomant | – Blocks GH receptors at target tissues |
- Prevents IGF-1 production
- Especially useful when GH levels are not controlled by other drugs
- Does not shrink tumor, so used in combination if tumor is present |
✅ 3. Dopamine Agonists (Adjunct Therapy)
| Drug | Action |
|---|---|
| Cabergoline, Bromocriptine | – Suppress GH secretion in some tumors |
- More effective if the tumor also secretes prolactin
- Oral agents, but generally less effective alone for gigantism |
✅ 4. Supportive/Adjunctive Care
- Antihypertensive drugs – for managing blood pressure
- Insulin or oral hypoglycemics – for managing insulin resistance or diabetes
- Nutritional counseling and physical therapy – for joint pain and bone support
🛠️ II. SURGICAL MANAGEMENT
🎯 Surgery is the preferred treatment for GH-secreting pituitary adenomas causing gigantism.
🔪 1. Transsphenoidal Adenomectomy
- Standard surgical procedure to remove pituitary tumors
- Performed through the nasal passage and sphenoid sinus
- First-line treatment if the tumor is accessible and operable
✅ Benefits:
- Can lead to immediate decrease in GH and IGF-1 levels
- Preserves normal pituitary function if successful
🛑 Risks:
- CSF leak
- Hypopituitarism
- Diabetes insipidus
- Incomplete tumor removal (in large or invasive tumors)
🔄 2. Repeat Surgery or Debulking
- Performed if:
- Tumor recurs
- Incomplete removal in the first surgery
- Rapid GH rebound post-treatment
💡 3. Radiation Therapy (Adjuvant/Second-Line)
| Type | Use |
|---|---|
| Conventional fractionated radiation | Given over weeks-months; slow response |
| Stereotactic radiosurgery (e.g., Gamma Knife) | Focused radiation; used when surgery is incomplete or contraindicated |
⚠️ May take months to years to reduce hormone levels
⚠️ Long-term risk of hypopituitarism
🩺 NURSING MANAGEMENT OF GIGANTISM
🎯 Goals of Nursing Care:
- Monitor and manage hormonal imbalances
- Support postoperative recovery and medication adherence
- Prevent or detect neurological and metabolic complications
- Provide psychological and developmental support
- Educate family and patient about lifelong care and follow-up
🗂️ I. Nursing Assessment
| Focus Area | Key Points |
|---|---|
| Growth and development | Monitor height, weight, bone age, pubertal status |
| Vital signs | Observe for hypertension, pulse irregularities |
| Vision assessment | Monitor for visual field defects (optic chiasm compression) |
| Neurological assessment | Watch for headache, behavior changes, signs of raised ICP |
| Musculoskeletal | Assess for joint pain, kyphosis, scoliosis |
| Psychosocial | Evaluate for low self-esteem, peer difficulties, or anxiety |
📝 II. Common Nursing Diagnoses
- Disturbed body image related to physical overgrowth
- Chronic pain related to joint/muscle strain
- Risk for injury related to impaired mobility or visual field changes
- Ineffective coping related to chronic illness or abnormal appearance
- Knowledge deficit regarding disease, treatment, and follow-up care
- Risk for impaired growth and development related to hormonal imbalance
🧾 III. Nursing Interventions
🔹 A. Preoperative Care (If Surgery Planned):
| Intervention | Purpose |
|---|---|
| Prepare child and family for transsphenoidal surgery | Reduce fear and increase cooperation |
| Monitor neurological signs, vision, and hormone levels | Establish baseline for comparison |
| Encourage psychological counseling/support groups | Help manage body image and emotional issues |
🔹 B. Postoperative Care:
| Intervention | Purpose |
|---|---|
| Monitor for CSF leak (clear nasal discharge), meningitis signs | Prevent life-threatening infections |
| Assess for diabetes insipidus | Monitor urine output and serum sodium levels |
| Monitor for hypopituitarism | Watch for fatigue, hypotension, growth failure post-op |
| Administer hormone replacements as prescribed | Prevent adrenal, thyroid, or gonadal insufficiency |
| Encourage rest and gradual activity | Prevent fatigue and reduce ICP risk |
🔹 C. Long-term Nursing Care:
| Intervention | Purpose |
|---|---|
| Educate on medication adherence (e.g., somatostatin analogs) | Promote hormone balance and tumor control |
| Schedule and reinforce regular follow-ups | Monitor IGF-1, GH levels, MRI imaging |
| Support nutritional and orthopedic management | Ensure joint protection and healthy weight |
| Promote school reintegration and peer acceptance | Help with developmental and social challenges |
| Provide family-centered education | Teach disease understanding, emergency signs, and home care routines |
📊 IV. Evaluation Criteria (Expected Outcomes):
- Patient maintains stable GH and IGF-1 levels
- Shows normal or slowed growth velocity
- Demonstrates improved self-esteem and coping skills
- Verbalizes understanding of medication regimen and follow-up care
- Remains free of postoperative or therapy-related complications
⚠️ COMPLICATIONS OF GIGANTISM
If left untreated or diagnosed late, gigantism can result in serious, sometimes lifelong complications due to prolonged GH and IGF-1 elevation, and structural abnormalities:
🧠 1. Neurological Complications
- Headaches, due to tumor pressure
- Bitemporal hemianopia from optic chiasm compression
- Hydrocephalus (rare, if tumor is large and obstructive)
💓 2. Cardiovascular Complications
- Hypertension
- Left ventricular hypertrophy → cardiomyopathy
- Heart failure (a leading cause of death in untreated cases)
🍬 3. Metabolic Complications
- Insulin resistance and Type 2 Diabetes Mellitus
- Hyperlipidemia
- Obesity (in some cases)
🦴 4. Musculoskeletal Problems
- Joint pain and early-onset osteoarthritis
- Kyphosis, scoliosis, and bone deformities
- Delayed closure of epiphyses → risk of fractures
🫁 5. Respiratory Complications
- Obstructive sleep apnea due to enlarged tongue, soft tissues
- Reduced lung function due to chest wall deformity
🧬 6. Endocrine/Reproductive Issues
- Delayed puberty, infertility, or menstrual irregularities
- Hypopituitarism after surgery or radiation
- Galactorrhea if prolactin is elevated
🧠 7. Psychological & Social Impact
- Depression, anxiety, low self-esteem due to appearance
- Social withdrawal, bullying in adolescence
- Academic issues due to vision or neurological effects
🧷 KEY POINTS ON GIGANTISM
✅ Definition: Overproduction of GH before epiphyseal plate closure → abnormally tall stature and tissue overgrowth
✅ Cause: Most often due to GH-secreting pituitary adenoma in children/adolescents
✅ Classic features:
- Rapid growth
- Enlarged hands, feet, facial features
- Delayed puberty, vision changes
✅ Diagnosis:
- ↑ IGF-1, failure of GH suppression in OGTT
- MRI of pituitary gland to locate tumor
✅ Treatment:
- Transsphenoidal surgery to remove pituitary tumor
- Somatostatin analogs, GH receptor antagonists, dopamine agonists
- Radiotherapy in resistant cases
✅ Nursing role:
- Monitor growth, glucose, BP, vision
- Post-op care: watch for CSF leak, DI, hypopituitarism
- Provide psychosocial support, promote adherence and education
✅ Complications:
- Cardiovascular disease, diabetes, skeletal deformities, infertility, psychosocial challenges
🧠 PROLACTINOMA
📌 Definition:
Prolactinoma is a benign pituitary tumor that overproduces prolactin, the hormone responsible for breast development and milk production. It is the most common type of pituitary adenoma and can affect both females and males, with symptoms varying by sex and age.
🔺 High prolactin levels (hyperprolactinemia) suppress reproductive hormones → infertility, amenorrhea, and galactorrhea.
🧬 Causes:
| Type | Causes |
|---|---|
| Primary | – Prolactin-secreting pituitary adenoma (prolactinoma) |
- Pituitary hyperplasia | | Secondary (Non-tumoral) | – Hypothyroidism
- Chest wall trauma
- Renal failure
- Pregnancy
- Certain drugs (e.g., antipsychotics, antidepressants, antiemetics, estrogen, methyldopa) |
🏷️ Types of Prolactinomas:
| Type | Description |
|---|---|
| Microprolactinoma | <10 mm in size; more common in females; may present with subtle hormonal symptoms |
| Macroprolactinoma | ≥10 mm in size; more common in males; can cause mass effect symptoms like headache and visual disturbances |
| Functional | Secretes active prolactin hormone |
| Non-functional pituitary tumor | May compress pituitary stalk → stalk effect → increased prolactin (not true prolactinoma) |
🔬 Pathophysiology:
- A pituitary adenoma develops from lactotroph cells in the anterior pituitary.
- These tumors secrete excessive prolactin.
- High prolactin:
- Inhibits GnRH → ↓ FSH and LH → ↓ estrogen/testosterone
- Disrupts ovarian and testicular function
- Stimulates milk production (even in non-pregnant females and males)
- Large tumors may compress surrounding tissues → optic chiasm compression, hypopituitarism
🚨 Signs and Symptoms:
Vary by tumor size and biological sex.
👩 In Women:
- Galactorrhea (milk discharge unrelated to childbirth)
- Amenorrhea or oligomenorrhea
- Infertility
- Decreased libido
- Vaginal dryness, dyspareunia
- Osteoporosis (due to low estrogen)
👨 In Men:
- Impotence, decreased libido
- Infertility
- Gynecomastia
- Galactorrhea (less common)
- Reduced muscle mass, fatigue
⚠️ Mass Effect Symptoms (in macroadenomas):
- Headache
- Visual field defects (especially bitemporal hemianopia)
- Hypopituitarism (fatigue, hypotension, loss of secondary sexual characteristics)
🧪 Diagnosis:
| Test | Findings |
|---|---|
| Serum prolactin | Markedly elevated (>200 ng/mL suggests prolactinoma) |
| TSH and FT4 | To rule out hypothyroidism (which can raise prolactin) |
| MRI brain – pituitary | Confirms tumor size and location |
| Visual field testing | For optic chiasm compression (macroadenomas) |
| Other pituitary hormones | Evaluate for hypopituitarism |
💊 Medical Management:
🎯 First-line treatment for prolactinoma is pharmacological, not surgery.
✅ 1. Dopamine Agonists (Gold Standard)
| Drug | Action |
|---|---|
| Cabergoline | First choice: more effective, fewer side effects |
| Bromocriptine | Alternative; safe in pregnancy |
🎯 These drugs lower prolactin levels, shrink tumors, and restore gonadal function.
✅ 2. Hormonal Support (if needed)
- Oral contraceptives or estrogen/testosterone replacement for hypogonadism
- Calcium and vitamin D for bone health
✅ 3. Regular Monitoring
- Serum prolactin levels every 3–6 months
- Repeat MRI annually for macroadenomas
- Visual field testing as indicated
🛠️ Surgical Management:
🎯 Reserved for patients who:
- Do not respond to or cannot tolerate dopamine agonists
- Have rapid tumor growth
- Experience vision loss or apoplexy
🔪 Transsphenoidal Surgery
- Minimally invasive surgery via nasal passage
- Removes tumor from pituitary
- Post-op: monitor for CSF leak, DI, hormone deficiencies
🩺 Nursing Management:
| Focus Area | Interventions |
|---|---|
| Monitoring | Vitals, vision changes, signs of hormone imbalance |
| Medication compliance | Ensure regular intake of dopamine agonists |
| Assess for side effects | Nausea, dizziness (from dopamine agonists) |
| Post-op care (if surgery) | Monitor nasal drainage (CSF leak), DI symptoms, neuro checks |
| Emotional support | Coping with infertility, body image, chronic disease |
| Patient education | Medication adherence, need for follow-up MRI/hormone testing, contraception during treatment (unless pregnancy is desired) |
⚠️ Complications:
- Vision loss from tumor growth
- Infertility (reversible with treatment)
- Bone demineralization from prolonged low estrogen/testosterone
- Hypopituitarism (after surgery or tumor compression)
- Pituitary apoplexy (sudden hemorrhage/infarction)
- Medication side effects (nausea, orthostatic hypotension, psychiatric effects)
🧷 Key Points on Prolactinoma
✅ Most common pituitary tumor, especially in women of reproductive age
✅ Caused by prolactin-secreting pituitary adenoma
✅ Symptoms: amenorrhea, galactorrhea, infertility, low libido
✅ Diagnosed via ↑ serum prolactin and MRI
✅ Dopamine agonists (cabergoline, bromocriptine) are first-line therapy
✅ Surgery is reserved for resistant or complicated tumors
✅ Nurses play a key role in medication adherence, monitoring, and supportive care
💧 Diabetes Insipidus (DI)
📌 Definition:
Diabetes Insipidus (DI) is a rare disorder characterized by the excretion of large volumes of dilute urine (polyuria) and intense thirst (polydipsia) due to either a deficiency of antidiuretic hormone (ADH, also called vasopressin) or the inability of the kidneys to respond to ADH. Unlike diabetes mellitus, DI is not related to glucose metabolism.
ADH is normally secreted by the posterior pituitary gland and acts on the kidneys to conserve water. In DI, this mechanism fails, leading to excessive water loss and a risk of dehydration, electrolyte imbalance, and hypovolemia.
🧬 Causes:
The causes of DI can be divided into central (neurogenic), nephrogenic, gestational, and behavioral (dipsogenic) forms:
- Central Diabetes Insipidus results from damage to the hypothalamus or posterior pituitary gland, leading to insufficient production or release of ADH. This damage may be due to:
- Head injury or trauma
- Neurosurgery (e.g., pituitary surgery)
- Brain tumors (pituitary adenoma, craniopharyngioma)
- Infections like meningitis or encephalitis
- Autoimmune inflammation
- Genetic mutations (rare)
- Idiopathic (no identifiable cause)
- Nephrogenic Diabetes Insipidus occurs when the kidneys are resistant to the action of ADH, even though it is present in normal or high amounts. Causes include:
- Chronic kidney disease
- Long-term use of certain drugs (especially lithium or demeclocycline)
- Electrolyte imbalances such as hypercalcemia and hypokalemia
- Congenital defects (mutations in ADH receptor or aquaporin-2 water channel genes)
- Gestational Diabetes Insipidus is a temporary form of DI that occurs during pregnancy, typically in the third trimester. It is caused by the placenta producing an enzyme called vasopressinase, which breaks down ADH. This form usually resolves after delivery.
- Dipsogenic Diabetes Insipidus, also called primary polydipsia, is caused by excessive water intake, which suppresses natural ADH secretion. It may occur in individuals with psychiatric disorders (such as schizophrenia) or damage to the hypothalamic thirst-regulating center.
🏷️ Types of Diabetes Insipidus:
- Central (Neurogenic) DI – Due to deficient production or secretion of ADH
- Nephrogenic DI – Due to renal resistance to ADH
- Gestational DI – Due to ADH degradation by placental enzymes during pregnancy
- Dipsogenic DI – Due to compulsive or habitual water drinking, leading to ADH suppression
🧬 Pathophysiology:
The central feature of diabetes insipidus is the inability of the body to conserve water, leading to excessive loss of dilute urine (polyuria) and resulting in intense thirst (polydipsia).
Under normal circumstances, antidiuretic hormone (ADH), also called vasopressin, is released by the posterior pituitary gland in response to dehydration or high plasma osmolality. ADH acts on the renal collecting ducts, increasing their permeability to water, which allows water to be reabsorbed into the bloodstream and concentrates the urine.
In central DI, there is a deficiency in ADH production or release, often due to damage to the hypothalamus or pituitary gland. As a result, water is not reabsorbed by the kidneys, leading to excessive loss of water in urine.
In nephrogenic DI, ADH is produced in adequate amounts, but the kidneys do not respond to it. This leads to the same outcome: inability to concentrate urine and loss of free water.
In gestational DI, an enzyme produced by the placenta (vasopressinase) degrades ADH, reducing its effectiveness. In dipsogenic DI, excessive fluid intake suppresses the release of ADH, mimicking the symptoms of true DI.
As the kidneys fail to concentrate urine, patients develop polyuria (usually >3 L/day in adults), leading to dehydration, increased thirst, low urine osmolality, and in severe cases, electrolyte imbalances.
🚨 Signs and Symptoms:
The hallmark symptoms of DI are:
- Polyuria: Excessive production of dilute urine (may exceed 5–10 liters per day in adults)
- Polydipsia: Intense, unquenchable thirst (especially for cold water)
- Nocturia: Frequent urination at night
- Dehydration: Dry skin, dry mouth, dizziness, and fatigue
- Weight loss: Due to fluid loss
- Low urine specific gravity: <1.005
- Hypotension and tachycardia: In severe dehydration
- Constipation or dry mucous membranes: In chronic cases
- Hypernatremia (↑ serum sodium): When water intake is inadequate to match losses
In infants or young children:
- Irritability
- Poor feeding
- Vomiting
- Fever
- Failure to thrive
- Seizures (due to electrolyte imbalance)
🧪 Diagnosis:
- History and Clinical Examination:
- History of excessive thirst and urination
- Evaluation of risk factors: head trauma, surgery, medications, pregnancy
- Laboratory Tests:
- Serum sodium: Often elevated (hypernatremia)
- Serum osmolality: High (>295 mOsm/kg)
- Urine osmolality: Low (<300 mOsm/kg)
- Urine specific gravity: Low (<1.005)
- 24-hour urine volume: Confirms polyuria (>3 liters/day in adults)
- Water Deprivation Test:
- Diagnostic gold standard
- Patient is restricted from fluids for several hours under supervision
- If urine remains dilute despite dehydration, it confirms DI
- Desmopressin (DDAVP) is administered:
- If urine becomes concentrated after DDAVP → Central DI
- If no change → Nephrogenic DI
- ADH (Vasopressin) Level:
- May be low in central DI
- Normal or high in nephrogenic DI
- Imaging (MRI brain):
- Used to evaluate pituitary or hypothalamic lesions, especially in central DI
💊 Medical Management:
The treatment of diabetes insipidus depends on its type (central, nephrogenic, gestational, or dipsogenic) and the underlying cause. The primary goals are to:
- Control fluid balance
- Reduce urine output
- Normalize serum electrolytes
- Treat the underlying disorder
✅ 1. Central Diabetes Insipidus:
- Desmopressin (DDAVP):
- Synthetic analog of ADH
- Drug of choice for central DI
- Administered intranasally, orally, or parenterally
- Reduces urine output and restores normal fluid balance
- Chlorpropamide or Carbamazepine:
- Occasionally used in mild cases to stimulate ADH release
- Rarely used now due to better options
- Fluids:
- Encourage oral fluids to prevent dehydration
- IV fluids (e.g., D5W) in cases of severe dehydration or unconsciousness
✅ 2. Nephrogenic Diabetes Insipidus:
- Thiazide diuretics (e.g., hydrochlorothiazide):
- Paradoxically reduce polyuria by inducing mild volume depletion
- Promotes proximal reabsorption of sodium and water
- Low-sodium, low-protein diet:
- Helps reduce urinary solute load
- Indomethacin or NSAIDs:
- Reduce urine volume by decreasing renal prostaglandin synthesis
- Treat underlying causes:
- Discontinue causative drugs like lithium
- Correct electrolyte imbalances (e.g., hypokalemia, hypercalcemia)
✅ 3. Gestational Diabetes Insipidus:
- Desmopressin (DDAVP):
- Safe and effective in pregnancy
- Not degraded by placental vasopressinase enzyme
- Symptoms usually resolve postpartum
✅ 4. Dipsogenic Diabetes Insipidus:
- Behavioral therapy:
- Patient education and fluid restriction under monitoring
- Treatment of underlying psychiatric disorder (if present)
🛠️ Surgical Management:
Surgery is not a primary treatment for most types of diabetes insipidus but may be needed in specific cases where the underlying cause is a structural lesion.
✅ Surgical Indications:
- Pituitary or hypothalamic tumor (e.g., craniopharyngioma, adenoma):
- May require transsphenoidal surgery
- Often followed by radiotherapy
- Postoperative patients may develop central DI and need lifelong desmopressin
- Traumatic brain injury with pituitary damage:
- If surgical decompression or CSF drainage is needed
- Hydrocephalus or brain malformations:
- Neurosurgical correction can relieve pressure and restore ADH function in some cases
⚠️ Post-Surgical Considerations:
- Close monitoring for:
- Polyuria or polydipsia
- Electrolyte imbalances (especially sodium)
- Signs of hypopituitarism
- Lifelong hormone replacement therapy may be required after pituitary surgery
🩺 NURSING MANAGEMENT OF DIABETES INSIPIDUS
🎯 Nursing Goals:
- Monitor and maintain fluid and electrolyte balance
- Prevent dehydration and hypovolemic shock
- Promote adherence to medications and dietary modifications
- Provide education about disease management and complications
- Support emotional and psychological well-being
🗂️ I. Assessment
- Monitor Intake and Output (I&O):
- Record urine output hourly (may exceed 200–500 mL/hour)
- Monitor for signs of polyuria and polydipsia
- Vital Signs Monitoring:
- Observe for hypotension, tachycardia, weak pulse (signs of dehydration)
- Assess for Dehydration:
- Dry mucous membranes
- Poor skin turgor
- Thirst and weakness
- Weight loss
- Monitor Neurological Status:
- Altered sensorium due to hypernatremia or fluid deficit
- Daily Weights:
- Early indicator of fluid imbalance
- Laboratory Monitoring:
- Serum sodium and osmolality
- Urine specific gravity (usually <1.005)
- Blood glucose (to rule out diabetes mellitus)
🧾 II. Nursing Interventions
🔹 A. Fluid Management
- Encourage oral fluids (preferably water) to prevent dehydration
- Administer IV fluids (e.g., D5W or hypotonic saline) as ordered in cases of severe dehydration
- Adjust fluid intake according to urine output
🔹 B. Medication Administration
- Administer Desmopressin (DDAVP) as prescribed (oral, nasal spray, or IV)
- Teach proper nasal spray technique if used at home
- Administer thiazide diuretics or NSAIDs for nephrogenic DI under supervision
- Monitor for side effects of all medications (e.g., hyponatremia due to over-treatment)
🔹 C. Nutrition and Electrolyte Balance
- Monitor and correct electrolyte imbalances (e.g., sodium, potassium)
- Provide a low-sodium, low-protein diet for nephrogenic DI patients if advised
- Educate patient/family on hydration and dietary needs
📘 III. Patient and Family Education
- Explain the nature of DI, its chronic course, and treatment options
- Teach the importance of strict fluid balance monitoring
- Instruct how to recognize early signs of dehydration or fluid overload
- Educate about lifelong medication adherence (especially in central DI)
- Stress the importance of regular follow-up visits and lab monitoring
🤝 IV. Emotional and Psychosocial Support
- Provide emotional reassurance and counseling
- Address body image, fatigue, or lifestyle adjustment issues
- Encourage participation in support groups, especially for chronic or genetic cases
📊 Evaluation (Expected Outcomes):
- Maintains adequate hydration and stable vital signs
- Exhibits normal serum sodium and osmolality
- Demonstrates understanding of disease and self-care practices
- Adheres to medication regimen and follow-up schedule
- Remains free from complications like hypovolemic shock or seizures
⚠️ COMPLICATIONS OF DIABETES INSIPIDUS
If untreated or poorly managed, DI can lead to serious and potentially life-threatening complications due to excessive fluid loss and electrolyte imbalance:
💧 1. Dehydration
- Caused by massive water loss through dilute urine
- Signs: dry skin, sunken eyes, low BP, dry mucosa, fatigue, dizziness
🧠 2. Hypernatremia
- Excessive water loss leads to elevated serum sodium levels
- May cause:
- Confusion
- Seizures
- Irritability
- Coma (in severe cases)
🫀 3. Hypovolemic Shock
- Sudden drop in blood pressure due to fluid depletion
- Life-threatening emergency
- Signs: tachycardia, hypotension, cold extremities
🧪 4. Electrolyte Imbalances
- Hypernatremia, hypokalemia, or hypercalcemia
- Can lead to cardiac arrhythmias or neuromuscular dysfunction
🧬 5. Growth and Development Issues (in children)
- Poor growth or failure to thrive
- Developmental delays if chronic DI is unrecognized
🧱 6. Complications of Treatment
- Water intoxication or hyponatremia due to over-treatment with desmopressin
- Nasal irritation or headache with DDAVP nasal spray
- GI upset or hypotension from thiazide diuretics or NSAIDs
📌 KEY POINTS ON DIABETES INSIPIDUS
✅ Definition: A disorder characterized by excessive urine output (polyuria) and intense thirst (polydipsia) due to ADH deficiency or renal resistance to ADH.
✅ Main Types:
- Central DI – due to ↓ ADH secretion
- Nephrogenic DI – due to renal resistance to ADH
- Gestational DI – due to placental destruction of ADH
- Dipsogenic DI – due to excess fluid intake suppressing ADH
✅ Key Symptoms:
- Polyuria, polydipsia, nocturia
- Dilute urine, low specific gravity
- Signs of dehydration and hypernatremia
✅ Diagnosis:
- Low urine osmolality, high serum osmolality
- Water deprivation test + Desmopressin response
- MRI to assess pituitary or hypothalamic abnormalities
✅ Treatment:
- Desmopressin for central and gestational DI
- Thiazide diuretics and low-sodium diet for nephrogenic DI
- Behavioral therapy for dipsogenic DI
✅ Nursing Role:
- Monitor I&O, hydration, vitals
- Educate on medication use and follow-up
- Prevent complications like dehydration and electrolyte imbalance
✅ Complications:
- Hypernatremia, dehydration, shock, seizures, growth issues
💦 SIADH – Syndrome of Inappropriate Antidiuretic Hormone Secretion
📌 Definition:
SIADH is a condition characterized by the excessive release of antidiuretic hormone (ADH) despite normal or low plasma osmolality, leading to water retention, hyponatremia (low serum sodium), and dilutional hypo-osmolality.
In SIADH, the kidneys reabsorb too much water, causing:
- Decreased urine output
- Dilute blood (low serum osmolality)
- Concentrated urine
🔁 “Too much ADH = Too much water retained = Dilutional hyponatremia”
🧬 Causes of SIADH:
SIADH may result from various conditions that cause abnormal ADH production or enhance its effect. Causes are grouped into central, pulmonary, malignancy-related, drug-induced, and other categories:
🧠 1. Central Nervous System (CNS) Disorders
- Head trauma
- Brain tumors (especially pituitary or hypothalamic)
- Meningitis, encephalitis
- Stroke, subarachnoid hemorrhage
- Post-neurosurgery
🫁 2. Pulmonary Disorders
- Pneumonia (especially viral or bacterial)
- Tuberculosis
- Acute respiratory failure
- Mechanical ventilation
- Positive pressure ventilation (stimulates ADH)
🧬 3. Malignancies (Paraneoplastic Syndrome)
Certain cancers can produce ectopic ADH, especially:
- Small cell lung carcinoma (most common)
- Pancreatic cancer
- Prostate and bladder cancer
- Leukemia and lymphoma
💊 4. Drug-Induced SIADH
Many medications can stimulate ADH release or enhance its action:
- Antidepressants: SSRIs (e.g., fluoxetine), tricyclics
- Antipsychotics: Haloperidol
- Antiepileptics: Carbamazepine, valproic acid
- Chemotherapy: Vincristine, cyclophosphamide
- Desmopressin, NSAIDs, opiates
🔍 5. Other Causes
- Pain, stress, or surgery
- Postoperative states
- Hypothyroidism or adrenal insufficiency (rare mimics)
🧾 Types of SIADH (Based on Cause):
| 🏷️ Type | 🔎 Description |
|---|---|
| Neurogenic SIADH | Due to CNS disorders (e.g., trauma, tumor, infection) |
| Pulmonary SIADH | Due to lung diseases like pneumonia or TB |
| Ectopic SIADH | ADH produced by tumors (e.g., small cell lung carcinoma) |
| Drug-induced SIADH | Medications increase ADH release or effect (e.g., SSRIs, anticonvulsants) |
| Idiopathic SIADH | No identifiable cause (often in elderly or chronically ill) |
📍 Quick Clinical Clue:
SIADH = Soaked Inside!
Excess ADH → Retains Water → Low Sodium → Concentrated Urine
🧬 Pathophysiology of SIADH:
In SIADH, the body secretes too much antidiuretic hormone (ADH) without a physiological need, which leads to:
- Excessive water reabsorption from the kidney’s collecting ducts
→ via action of ADH on V2 receptors - Dilution of plasma (water retained but not sodium)
→ resulting in hypo-osmolality and hyponatremia - Urine becomes concentrated
→ because sodium and water are still excreted together initially - As serum sodium drops, brain cells swell due to osmotic imbalance
→ leading to cerebral edema and neurological symptoms
⚠️ Key Outcome:
Water retained inside → Low sodium, low serum osmolality, high urine osmolality
🚨 Signs and Symptoms of SIADH:
Symptoms primarily reflect hyponatremia severity and cerebral edema:
💧 Early/Mild Symptoms (Na+ 130–135 mEq/L):
- Nausea
- Loss of appetite
- Headache
- Mild fatigue
- Muscle cramps
🧠 Moderate Symptoms (Na+ 120–129 mEq/L):
- Lethargy
- Disorientation
- Irritability
- Muscle twitching
- Confusion
- Slowed reflexes
⚠️ Severe Symptoms (Na+ <120 mEq/L):
- Seizures
- Delirium
- Coma
- Decreased consciousness
- Respiratory arrest (late stage, brainstem herniation)
🧠 Neurological symptoms are due to cerebral edema from water shifting into brain cells.
🧪 Diagnosis of SIADH:
SIADH is a diagnosis of exclusion. The following labs and assessments help confirm the condition:
✅ Laboratory Investigations:
| 🔬 Test | 🔍 Finding in SIADH |
|---|---|
| Serum sodium (Na⁺) | ↓ Low (<135 mEq/L) → hyponatremia |
| Serum osmolality | ↓ Low (<275 mOsm/kg) → dilutional effect |
| Urine osmolality | ↑ High (>100 mOsm/kg) → concentrated despite low serum osmolality |
| Urine sodium | ↑ High (>40 mEq/L) → inappropriate sodium loss |
| Serum urea, creatinine, uric acid | ↓ Decreased due to hemodilution |
| Serum potassium | Usually normal |
🩺 Other Diagnostic Workup:
- Thyroid function tests (TSH, T4) → to rule out hypothyroidism
- Cortisol level → to exclude adrenal insufficiency (Addison’s disease)
- Chest X-ray / CT scan → to check for lung tumors or pneumonia
- MRI brain → to rule out CNS disorders (e.g., tumor, trauma)
📍 Diagnostic Criteria (Bartter & Schwartz Criteria):
- Hyponatremia with low plasma osmolality
- Inappropriately concentrated urine (not maximally dilute)
- Euvolemia (no signs of dehydration or fluid overload)
- Normal renal, thyroid, and adrenal function
- No diuretic use
💊 Medical Management of SIADH
The primary goals of treatment are to:
✅ Correct hyponatremia
✅ Manage fluid retention
✅ Treat the underlying cause (e.g., tumor, infection, drug effect)
🔹 1. Fluid Restriction (First-line management)
- Restrict fluids to 800–1000 mL/day
- Effective in mild to moderate hyponatremia
- Educate patient to avoid free water intake (e.g., juices, soup, coffee)
- Strict input and output (I&O) monitoring is essential
🔹 2. Salt Supplementation
- Oral salt tablets or hypertonic saline (3% NaCl) may be given in:
- Severe hyponatremia (Na+ <120 mEq/L)
- Neurological symptoms like seizures or altered consciousness
- Must be administered slowly to avoid central pontine myelinolysis
🔹 3. Loop Diuretics (e.g., Furosemide)
- Used to increase free water clearance
- Given along with saline infusion to prevent worsening of hyponatremia
- Monitor electrolytes regularly
🔹 4. Vasopressin Receptor Antagonists (Vaptans)
| Drug | Action |
|---|---|
| Tolvaptan (oral) | |
| Conivaptan (IV) | – Block V2 receptors in the kidneys |
- Promote water excretion without losing sodium
- Used in moderate to severe cases or if fluid restriction fails
⚠️ Use with caution — may cause rapid correction of sodium
Requires hospital monitoring
🔹 5. Address Underlying Causes
- Discontinue causative drugs (e.g., SSRIs, carbamazepine, vincristine)
- Treat CNS infections, trauma, tumors
- Manage lung infections (e.g., pneumonia, TB)
- Treat malignancies producing ectopic ADH (e.g., small cell lung cancer)
🛠️ Surgical Management of SIADH
Surgery is not directly performed to correct SIADH, but may be needed when:
✅ 1. Tumor Removal (Underlying Cause)
- Surgical excision of ectopic ADH-secreting tumors (e.g., small cell lung carcinoma, CNS tumors, pancreatic tumors)
- May lead to permanent resolution of SIADH
✅ 2. Neurosurgical Intervention
- Required in cases of pituitary or hypothalamic tumors, hydrocephalus, or traumatic brain injury
- Postoperative monitoring for fluid balance and sodium levels is essential
✅ 3. Ventilation Management
- For SIADH induced by mechanical ventilation (positive pressure can stimulate ADH)
- Adjusting ventilator settings and sedatives can help reduce ADH release
⚠️ Important Monitoring During Management:
- Daily weights
- Serum sodium and osmolality every 4–6 hours
- Watch for symptoms of overcorrection of sodium:
- Lethargy
- Confusion
- Muscle tremors
- Seizures
🩺 NURSING MANAGEMENT OF SIADH
🎯 Primary Nursing Goals:
- Monitor and manage fluid and electrolyte balance
- Prevent complications from hyponatremia
- Promote patient safety and neurological stability
- Educate the patient and family about fluid restriction and treatment
🗂️ I. Assessment and Monitoring:
🔸 Fluid Intake & Output (I&O):
- Maintain strict documentation of all oral and IV fluids
- Monitor for decreased urine output
🔸 Daily Weights:
- Weigh patient daily at the same time
- Sudden weight gain = fluid retention
🔸 Vital Signs Monitoring:
- Observe for hypertension, tachycardia, or hypothermia
- Assess for signs of fluid overload (edema, crackles)
🔸 Neurological Assessments:
- Monitor for changes in level of consciousness, confusion, headache, seizures
- Be vigilant with sodium levels <125 mEq/L
🔸 Laboratory Values:
- Monitor:
- Serum sodium (Hyponatremia <135 mEq/L)
- Serum osmolality (↓ <275 mOsm/kg)
- Urine osmolality (↑ >100 mOsm/kg)
- Urine sodium (↑ >40 mEq/L)
🧾 II. Nursing Interventions:
🔹 Fluid Restriction:
- Limit fluid intake to 800–1000 mL/day (or as prescribed)
- Offer ice chips, sugarless gum, or oral swabs for dry mouth
- Instruct family to avoid bringing extra drinks to the patient
🔹 Medication Administration:
- Administer diuretics, hypertonic saline, or vasopressin antagonists as prescribed
- Monitor for overcorrection of sodium (rapid rise may cause central pontine myelinolysis)
- Observe for side effects of medications (e.g., thirst, dizziness, GI upset)
🔹 Safety Measures:
- Implement seizure precautions (padded side rails, oxygen, suction setup)
- Assist with ambulation (risk of falls due to confusion, weakness)
🔹 Nutritional Support:
- Provide high-sodium diet (if prescribed)
- Educate on avoiding low-sodium foods during hyponatremia
- Monitor swallowing ability if neurological status is altered
📘 III. Patient and Family Education:
- Explain what SIADH is and why fluid restriction is important
- Teach early symptoms of hyponatremia: nausea, headache, confusion
- Emphasize importance of regular blood tests to monitor sodium
- Instruct to avoid over-the-counter medications that may worsen SIADH (e.g., NSAIDs, certain antidepressants)
📊 IV. Evaluation (Expected Outcomes):
✅ Maintains normal serum sodium levels
✅ Demonstrates neurological stability
✅ Adheres to fluid restriction and medication plan
✅ Verbalizes understanding of the condition and prevention strategies
✅ Remains free from complications like seizures or coma
⚠️ COMPLICATIONS OF SIADH
When SIADH is not identified or properly managed, it can lead to serious complications, primarily due to dilutional hyponatremia and cerebral edema.
🧠 1. Neurological Complications
- Cerebral edema (swelling of brain cells)
- Headache, confusion, irritability
- Seizures, coma, and even death in severe hyponatremia
- Brainstem herniation (rare but fatal if sodium drops too rapidly)
🧪 2. Electrolyte Imbalance
- Persistent hyponatremia (<120 mEq/L)
- May result in muscle cramps, weakness, or altered reflexes
- Hypokalemia if diuretics are used excessively
💉 3. Osmotic Demyelination Syndrome (Central Pontine Myelinolysis)
- Occurs if serum sodium is corrected too quickly
- Leads to permanent brain damage
- Symptoms: dysarthria, paralysis, coma
🫁 4. Fluid Overload
- If fluid restriction is not followed:
- Pulmonary edema
- Hypertension
- Heart failure in vulnerable patients
📌 KEY POINTS ON SIADH
✅ Definition: SIADH is the excess secretion of ADH, causing water retention, dilutional hyponatremia, and concentrated urine.
✅ Common Causes:
- CNS disorders (e.g., head injury, tumor, stroke)
- Lung diseases (e.g., pneumonia, TB)
- Small cell lung cancer (ectopic ADH secretion)
- Medications: SSRIs, anticonvulsants, chemo
✅ Key Features:
- Low serum sodium & osmolality
- High urine sodium & osmolality
- No signs of fluid overload (euvolemic hyponatremia)
✅ Major Symptoms:
- Early: nausea, headache, fatigue
- Moderate: confusion, irritability
- Severe: seizures, coma, respiratory arrest
✅ Diagnosis:
- Based on labs, clinical picture, and rule out of other causes (adrenal, thyroid, renal)
✅ Treatment:
- Fluid restriction (mainstay)
- Hypertonic saline (for severe cases)
- Loop diuretics, salt tablets, or vaptans
- Treat underlying cause (e.g., infection, tumor, drug)
✅ Nursing Focus:
- Monitor I&O, neuro signs, labs
- Prevent complications
- Educate on fluid restriction and medication adherence
✅ Complications:
- Seizures, coma, central pontine myelinolysis, fluid overload
🍬 Diabetes Mellitus (DM)
📌 Definition & Causes
✅ Definition:
Diabetes Mellitus is a chronic metabolic disorder characterized by hyperglycemia (elevated blood glucose levels) due to:
- Deficiency of insulin secretion,
- Resistance to insulin action, or
- Both.
Insulin is a hormone produced by the beta cells of the pancreas (Islets of Langerhans) that regulates glucose uptake into cells for energy. When insulin is inadequate or ineffective, glucose remains in the bloodstream, leading to persistent high blood sugar and disturbances in carbohydrate, fat, and protein metabolism.
🔍 Chronic uncontrolled diabetes can affect the eyes, kidneys, nerves, heart, and blood vessels.
🧬 Causes of Diabetes Mellitus:
The causes vary by the type of diabetes but generally involve genetic, autoimmune, environmental, and lifestyle factors.
🟠 1. Type 1 Diabetes Mellitus (T1DM):
- Autoimmune destruction of pancreatic beta cells → absolute insulin deficiency
- Usually occurs in children or young adults
- Can also be idiopathic (without a known cause)
Common Causes:
- Autoimmune reaction (anti-GAD antibodies)
- Genetic predisposition (HLA-DR3, DR4)
- Viral infections (e.g., Coxsackievirus, mumps)
- Family history of Type 1 DM
🔵 2. Type 2 Diabetes Mellitus (T2DM):
- Characterized by insulin resistance and relative insulin deficiency
- Most common type, often seen in adults, but increasingly in children due to obesity
Common Causes:
- Obesity, especially central (abdominal) obesity
- Physical inactivity
- Poor dietary habits (high-sugar, high-fat intake)
- Genetic predisposition
- Aging
- History of gestational diabetes or polycystic ovarian syndrome (PCOS)
- Ethnic background (e.g., South Asians, African Americans)
🟡 3. Gestational Diabetes Mellitus (GDM):
- Develops during pregnancy due to hormonal changes causing insulin resistance
- Usually resolves after delivery, but increases risk of future T2DM
Causes/Risk Factors:
- Hormones (placental lactogen, cortisol) interfere with insulin
- Obesity during pregnancy
- Family history of diabetes
- Previous macrosomic baby
- Advanced maternal age
🟢 4. Other Specific Causes (Secondary Diabetes):
- Endocrine disorders: Cushing’s syndrome, acromegaly, hyperthyroidism
- Pancreatic diseases: Pancreatitis, cystic fibrosis, hemochromatosis
- Drug-induced: Steroids, thiazides, antipsychotics
- Genetic defects: MODY (Maturity Onset Diabetes of the Young), mitochondrial mutations
🔢 Types of Diabetes Mellitus (DM)
Diabetes mellitus is classified into several types based on its cause, age of onset, and pathophysiology. The four major types are:
1️⃣ Type 1 Diabetes Mellitus (T1DM) – Insulin-Dependent Diabetes
- Caused by autoimmune destruction of pancreatic β-cells, leading to absolute insulin deficiency
- Usually occurs in childhood or adolescence, but can occur at any age
- Requires lifelong insulin therapy
Key Features:
- Rapid onset
- Weight loss despite increased appetite
- Polyuria, polydipsia, polyphagia
- Positive for autoantibodies (GAD, ICA)
- Risk of diabetic ketoacidosis (DKA)
2️⃣ Type 2 Diabetes Mellitus (T2DM) – Non-Insulin-Dependent Diabetes
- Caused by insulin resistance in body tissues and a relative insulin deficiency
- Most common type (≈90–95% of all diabetes cases)
- Often associated with obesity, sedentary lifestyle, and genetic predisposition
Key Features:
- Gradual onset
- May be asymptomatic for years
- Managed by diet, exercise, oral antidiabetics, and sometimes insulin
- Increased risk of cardiovascular disease and complications
3️⃣ Gestational Diabetes Mellitus (GDM)
- Glucose intolerance that develops during pregnancy, usually in the 2nd or 3rd trimester
- Caused by placental hormones (e.g., hPL, cortisol) that create insulin resistance
Key Features:
- Temporary but may recur in future pregnancies
- Increases risk of Type 2 DM later in life
- Associated with fetal complications (macrosomia, neonatal hypoglycemia)
4️⃣ Other Specific Types (Secondary Diabetes)
These are less common and occur due to specific causes:
🔹 A. Genetic Defects in β-cell Function:
- MODY (Maturity Onset Diabetes of the Young)
- Neonatal diabetes mellitus
🔹 B. Diseases of the Pancreas:
- Chronic pancreatitis
- Pancreatic cancer
- Hemochromatosis
🔹 C. Endocrinopathies:
- Cushing’s syndrome
- Acromegaly
- Pheochromocytoma
- Hyperthyroidism
🔹 D. Drug- or Chemical-Induced Diabetes:
- Long-term use of glucocorticoids
- Thiazide diuretics
- Antipsychotics
- Immunosuppressants (e.g., tacrolimus)
🔹 E. Infections:
- Congenital rubella
- Cytomegalovirus
🧬 Pathophysiology of Diabetes Mellitus
📌 Overview:
Diabetes mellitus is marked by a deficiency of insulin or the body’s inability to use insulin effectively, leading to elevated blood glucose levels (hyperglycemia). The metabolic consequences affect carbohydrate, fat, and protein metabolism.
🔷 Type 1 Diabetes Mellitus (T1DM) – Pathophysiology:
- Autoimmune reaction targets and destroys the β-cells in the pancreatic islets of Langerhans, where insulin is produced.
- This leads to absolute insulin deficiency → the body cannot use glucose for energy.
- Glucose accumulates in the bloodstream (hyperglycemia) and is excreted in urine (glycosuria), pulling water with it → polyuria and dehydration.
- As glucose cannot enter cells:
- The body uses fats for energy, producing ketone bodies → ketosis
- If unchecked, this leads to diabetic ketoacidosis (DKA) – a life-threatening emergency.
🔍 In T1DM, insulin is virtually absent, and patients are dependent on insulin injections for survival.
🔶 Type 2 Diabetes Mellitus (T2DM) – Pathophysiology:
- Begins with insulin resistance: body cells (muscle, fat, liver) fail to respond to insulin properly.
- The pancreas compensates by producing more insulin (hyperinsulinemia).
- Over time, β-cell dysfunction develops, and insulin secretion declines.
- The result is relative insulin deficiency and persistent hyperglycemia.
- Excess glucose in the bloodstream causes:
- Increased fat breakdown → dyslipidemia
- Endothelial damage → microvascular and macrovascular complications (retinopathy, nephropathy, neuropathy, atherosclerosis)
🔍 In T2DM, insulin is present but not effective—eventually requiring medication or insulin.
🔁 Common Effects in Both Types:
- Hyperglycemia → cellular dehydration
- Glycosuria → osmotic diuresis → polyuria and dehydration
- Polydipsia → due to fluid loss
- Polyphagia → due to glucose not entering cells
- Weight loss → due to fat/protein breakdown
- Chronic complications from glucose toxicity:
- Eyes (retinopathy)
- Kidneys (nephropathy)
- Nerves (neuropathy)
- Heart & vessels (coronary artery disease, stroke)
✅ Signs and Symptoms of Diabetes Mellitus
The classic symptoms result from persistent hyperglycemia, cellular glucose deprivation, and osmotic diuresis. Symptoms may vary slightly depending on Type 1, Type 2, or Gestational Diabetes.
🔷 Common Symptoms in Both Type 1 & Type 2:
- Polyuria – Frequent urination due to glucose-induced osmotic diuresis
- Polydipsia – Excessive thirst due to dehydration
- Polyphagia – Increased hunger (glucose not entering cells)
- Weight loss – Especially in Type 1, due to fat and protein breakdown
- Fatigue and weakness – Due to energy depletion
- Blurred vision – Due to fluid shifts in the eye lens
- Slow wound healing – High sugar impairs immune and tissue repair functions
- Tingling or numbness – Peripheral neuropathy in chronic diabetes
- Recurrent infections – Especially skin, urinary tract, or genital infections
🔶 Type 1 Diabetes Specific:
- Sudden onset of symptoms
- Common in children or adolescents
- More likely to present with diabetic ketoacidosis (DKA) (nausea, vomiting, fruity breath, abdominal pain, rapid breathing)
🔵 Type 2 Diabetes Specific:
- Gradual onset; may remain undiagnosed for years
- Often detected during routine blood tests
- Frequently associated with obesity, hypertension, or dyslipidemia
🟡 Gestational Diabetes Specific:
- Usually asymptomatic
- May present with:
- Excessive weight gain
- Increased thirst or urination
- Detected during routine prenatal screening
🧪 Diagnosis of Diabetes Mellitus
Diagnosis is based on blood glucose levels, either fasting, random, or after a glucose load, and HbA1c (glycated hemoglobin).
✅ 1. Fasting Blood Glucose (FBG):
- Normal: <100 mg/dL
- Diabetes: ≥126 mg/dL (after 8–10 hours fasting)
- Impaired Fasting Glucose (prediabetes): 100–125 mg/dL
✅ 2. Random Blood Glucose (RBG):
- Normal: <140 mg/dL
- Diabetes: ≥200 mg/dL with symptoms
✅ 3. Oral Glucose Tolerance Test (OGTT):
- 2-hour post 75g glucose load:
- Normal: <140 mg/dL
- Prediabetes: 140–199 mg/dL
- Diabetes: ≥200 mg/dL
✅ 4. HbA1c (Glycosylated Hemoglobin):
- Reflects average blood glucose over 2–3 months
- Normal: <5.7%
- Prediabetes: 5.7% – 6.4%
- Diabetes: ≥6.5%
🔍 Additional Tests:
- Urinalysis: Glucose and ketones
- C-peptide test: To differentiate Type 1 (low/absent) vs. Type 2 (normal/high)
- Autoantibody tests: (GAD, ICA) for Type 1 DM
- Lipid profile: For cardiovascular risk assessment
- Renal function tests: Urea, creatinine, microalbuminuria
- ECG or fundus examination: In chronic diabetes
💊 Medical Management of Diabetes Mellitus
🎯 Goals of Treatment:
- Achieve and maintain optimal blood glucose levels
- Prevent acute complications like hypoglycemia, DKA, or HHS
- Prevent or delay chronic complications (retinopathy, nephropathy, neuropathy)
- Improve overall quality of life
🔷 1. Management of Type 1 Diabetes Mellitus (T1DM):
Because T1DM involves absolute insulin deficiency, patients require lifelong insulin therapy.
✅ Insulin Therapy (Lifelong)
🔹 Types of Insulin:
- Rapid-acting (e.g., Lispro, Aspart): onset 15 min, peak 1 hr
- Short-acting (e.g., Regular): onset 30–60 min, peak 2–3 hrs
- Intermediate-acting (e.g., NPH): onset 2–4 hrs, peak 4–12 hrs
- Long-acting (e.g., Glargine, Detemir): minimal peak, lasts 24 hrs
🔹 Regimens:
- Basal-bolus therapy: Combines long-acting + rapid-acting insulins
- Insulin pumps: Provide continuous subcutaneous insulin infusion
- Sliding scale insulin: Used in hospitals based on blood sugar readings
🔹 Monitoring:
- Frequent blood glucose monitoring (4–6 times/day)
- HbA1c every 3 months
- Adjust insulin based on meals, stress, exercise
🔶 2. Management of Type 2 Diabetes Mellitus (T2DM):
Type 2 DM treatment begins with lifestyle changes, followed by oral antidiabetic drugs (OADs) and possibly insulin if needed.
✅ A. Lifestyle Modifications (Always First Step):
- Balanced low-sugar, low-fat diet
- Regular physical activity (30–45 minutes/day)
- Weight reduction (goal: BMI <25)
- Smoking and alcohol cessation
✅ B. Oral Antidiabetic Drugs (OADs):
| 🧪 Drug Class | 💊 Examples | 🧬 Mechanism of Action |
|---|---|---|
| Biguanides | Metformin | ↓ hepatic glucose production, ↑ insulin sensitivity |
| Sulfonylureas | Glipizide, Glyburide | ↑ insulin secretion from pancreas |
| Meglitinides | Repaglinide, Nateglinide | ↑ rapid insulin secretion (short-acting) |
| Thiazolidinediones | Pioglitazone, Rosiglitazone | ↑ insulin sensitivity in fat and muscle tissues |
| DPP-4 Inhibitors | Sitagliptin, Vildagliptin | ↑ incretin effect → ↑ insulin, ↓ glucagon |
| SGLT-2 Inhibitors | Empagliflozin, Dapagliflozin | ↑ renal glucose excretion |
| Alpha-glucosidase inhibitors | Acarbose, Miglitol | ↓ carbohydrate absorption in intestine |
🔍 Metformin is the first-line drug unless contraindicated (e.g., renal disease).
✅ C. Insulin in Type 2 Diabetes (if needed):
- Indicated when oral drugs fail, during infections, surgery, pregnancy, or very high blood glucose levels
- May use basal insulin at night, or intensive insulin therapy in advanced cases
🟡 3. Management of Gestational Diabetes Mellitus (GDM):
- Start with diet and exercise
- If glucose remains uncontrolled → insulin is preferred (safe in pregnancy)
- Oral drugs like metformin may be considered in selected cases
- Close fetal monitoring required
🧾 Other Medical Interventions (for all types):
- Statins: To control cholesterol
- ACE inhibitors/ARBs: For BP and kidney protection
- Low-dose aspirin: If cardiovascular risk is high
- Multivitamins and B-complex: For neuropathy support
- Foot care advice: To prevent diabetic foot ulcers
🛠️ Surgical Management of Diabetes Mellitus
🔷 A. Bariatric Surgery (Metabolic Surgery)
🎯 Used in obese patients with Type 2 Diabetes Mellitus (T2DM)
Can significantly improve or even resolve diabetes in many cases.
✅ Indications:
- BMI ≥ 40 kg/m² (without comorbidities)
- BMI ≥ 35 kg/m² with uncontrolled diabetes or comorbidities (e.g., hypertension, sleep apnea)
- Poor control of blood glucose despite medication and lifestyle modification
✅ Common Bariatric Procedures:
- Roux-en-Y Gastric Bypass (RYGB)
- Reduces stomach size and bypasses part of small intestine
- Decreases calorie absorption and improves insulin sensitivity
- Sleeve Gastrectomy
- Removes part of the stomach → reduces hunger hormone (ghrelin)
- Improves insulin response
- Adjustable Gastric Banding
- Silicone band placed around stomach to limit food intake
- Less effective for diabetes resolution
- Biliopancreatic Diversion with Duodenal Switch (BPD-DS)
- Most effective but also most complex
- Rarely done due to high risk of nutritional deficiencies
📊 Benefit: Many patients show complete or partial remission of Type 2 DM within months.
🔶 B. Pancreas Transplantation
🎯 Used in selected patients with Type 1 Diabetes Mellitus
Offers a potential cure by restoring insulin production.
✅ Indications:
- Type 1 diabetes with end-stage renal disease (ESRD)
- Combined kidney-pancreas transplant is common
- Severe hypoglycemia unawareness
- Recurrent diabetic ketoacidosis (DKA)
✅ Types of Transplant:
- Pancreas transplant alone (PTA)
- Simultaneous pancreas-kidney (SPK) transplant
- Pancreas after kidney (PAK) transplant
🔍 Lifelong immunosuppression is required post-transplant to prevent rejection.
🟡 C. Surgical Interventions for Diabetes Complications
Diabetes affects multiple organs and may require surgical management of:
1. Diabetic Foot Ulcers:
- Debridement of necrotic tissue
- Skin grafting
- Amputation (in severe, non-healing infected wounds)
2. Ophthalmic Complications:
- Laser photocoagulation for diabetic retinopathy
- Vitrectomy for vitreous hemorrhage or retinal detachment
3. Renal Complications:
- Renal transplantation in end-stage diabetic nephropathy
🧠 Summary:
| 🛠️ Surgery | 🩺 Purpose |
|---|---|
| Bariatric surgery | Weight loss & T2DM remission |
| Pancreas transplant | Type 1 DM cure in selected patients |
| Foot surgery | Infection control & limb preservation |
| Eye surgery | Prevent blindness from diabetic retinopathy |
| Renal transplant | ESRD due to diabetic nephropathy |
🩺 NURSING MANAGEMENT OF DIABETES MELLITUS
🎯 Goals of Nursing Care:
- Maintain normal or near-normal blood glucose levels
- Prevent acute complications (hypoglycemia, DKA, HHS)
- Prevent or delay chronic complications (retinopathy, nephropathy, neuropathy)
- Promote independence and lifestyle adjustment
- Enhance patient education and adherence to therapy
🗂️ I. Assessment and Monitoring
✅ Vital Signs:
- Monitor blood pressure (diabetes + hypertension is common)
- Monitor temperature (infection risk is high)
✅ Blood Glucose Monitoring:
- Check capillary blood glucose regularly (before meals/bedtime or as per doctor’s order)
- Observe for hypo/hyperglycemia symptoms
✅ Weight and BMI:
- Record baseline and periodic weight to assess lifestyle interventions
✅ Foot Inspection:
- Daily foot checks for ulcers, wounds, infections
- Monitor peripheral circulation and sensation (neuropathy signs)
✅ Lab Values:
- Fasting/random blood glucose
- HbA1c (every 3 months)
- Lipid profile, renal function, urine albumin
- Electrolytes in acute illness
🧾 II. Interventions and Nursing Actions
🔹 1. Medication Administration:
- Administer insulin or oral hypoglycemic agents as prescribed
- Verify timing with meals to prevent hypoglycemia
- Rotate insulin injection sites to avoid lipodystrophy
- Monitor for side effects of medications
🔹 2. Dietary Management:
- Coordinate with dietitian for a diabetic diet plan
- Educate patient on:
- Carbohydrate counting
- Low sugar, low fat, high fiber foods
- Consistent meal times and snacks to prevent hypoglycemia
🔹 3. Exercise and Activity:
- Encourage regular moderate exercise (30 minutes/day)
- Monitor for signs of hypoglycemia during activity
- Avoid exercise during uncontrolled hyperglycemia or ketonuria
🔹 4. Hydration:
- Encourage adequate fluid intake, especially in hyperglycemia
- Monitor for signs of dehydration (dry skin, sunken eyes, hypotension)
📘 III. Patient Education and Health Promotion
- Teach self-monitoring of blood glucose (SMBG)
- Educate about signs/symptoms of:
- Hypoglycemia: shakiness, sweating, hunger, confusion
- Hyperglycemia: thirst, dry mouth, frequent urination, blurred vision
- Instruct on foot care:
- Wash daily, dry well, inspect for cracks/cuts
- Never walk barefoot, wear proper shoes
- Emphasize importance of:
- Regular follow-up visits
- Eye check-ups (annually)
- Kidney and heart screenings
- Support stress reduction, as it may elevate blood glucose
⚠️ IV. Preventing and Managing Complications
- Hypoglycemia:
- Administer 15g quick-acting carbohydrates (e.g., juice, glucose tablets)
- Recheck sugar after 15 minutes
- Prepare to give glucagon IM or IV dextrose in severe cases
- Diabetic Ketoacidosis (DKA)/HHS:
- Monitor vitals, blood gases, ketones, hydration
- Prepare for IV fluids, insulin therapy, and electrolyte replacement
📊 V. Evaluation (Expected Outcomes):
✅ Maintains blood glucose within target range
✅ Verbalizes understanding of disease and management
✅ Demonstrates correct insulin/self-care technique
✅ Shows no signs of complications
✅ Adheres to diet, medication, and lifestyle changes
⚠️ COMPLICATIONS OF DIABETES MELLITUS
Diabetes, especially when poorly controlled, can lead to acute emergencies and chronic multi-organ damage.
🩸 A. Acute Complications
🔹 1. Hypoglycemia
- Occurs when blood glucose <70 mg/dL
- Causes: excess insulin, missed meals, over-exercise
- Symptoms: shakiness, sweating, confusion, blurred vision, seizures
- Can lead to coma or death if untreated
🔹 2. Diabetic Ketoacidosis (DKA) – Type 1 DM
- Due to absolute insulin deficiency → fat breakdown → ketone production
- Signs: fruity breath, vomiting, rapid breathing, abdominal pain
- Life-threatening; needs IV insulin and fluids
🔹 3. Hyperosmolar Hyperglycemic State (HHS) – Type 2 DM
- Very high blood sugar without ketones
- Severe dehydration and altered consciousness
- Requires aggressive rehydration and insulin
🧠 B. Chronic Complications
🔹 1. Microvascular Complications (small vessels)
- Diabetic Retinopathy → blindness
- Diabetic Nephropathy → kidney failure
- Diabetic Neuropathy → numbness, foot ulcers, infections
🔹 2. Macrovascular Complications (large vessels)
- Coronary artery disease (CAD) → heart attack
- Cerebrovascular disease → stroke
- Peripheral artery disease (PAD) → leg ulcers, gangrene
🔹 3. Diabetic Foot
- Neuropathy + poor circulation → foot ulcers → infection → amputation
🔹 4. Increased Risk of Infections
- Especially in the skin, urinary tract, respiratory system
- Delayed wound healing
📌 KEY POINTS ON DIABETES MELLITUS
✅ Definition: Chronic metabolic disorder with high blood sugar due to insulin deficiency or resistance
✅ Main Types:
- Type 1 DM – autoimmune, insulin-dependent
- Type 2 DM – lifestyle-related, insulin resistance
- Gestational DM – during pregnancy
- Secondary DM – due to diseases/drugs
✅ Classic Symptoms:
Polyuria, Polydipsia, Polyphagia, Weight loss, Fatigue
✅ Diagnosis:
- Fasting Glucose ≥126 mg/dL
- HbA1c ≥6.5%
- 2-hr OGTT ≥200 mg/dL
- Random Glucose ≥200 mg/dL with symptoms
✅ Medical Management:
- Type 1 → Insulin lifelong
- Type 2 → Lifestyle + Oral drugs ± Insulin
- Gestational → Diet ± Insulin
✅ Nursing Role:
- Monitor glucose, educate on insulin, foot care, diet
- Prevent complications like hypoglycemia, DKA, and infections
✅ Complications:
- Acute: Hypoglycemia, DKA, HHS
- Chronic: Eye, kidney, nerve, heart, foot damage