B.SC-PAEDIA-UNIT-5
Nursing management in
common childhood diseases.
Respiratory system:
Tracheoesophageal Fistula (TEF)
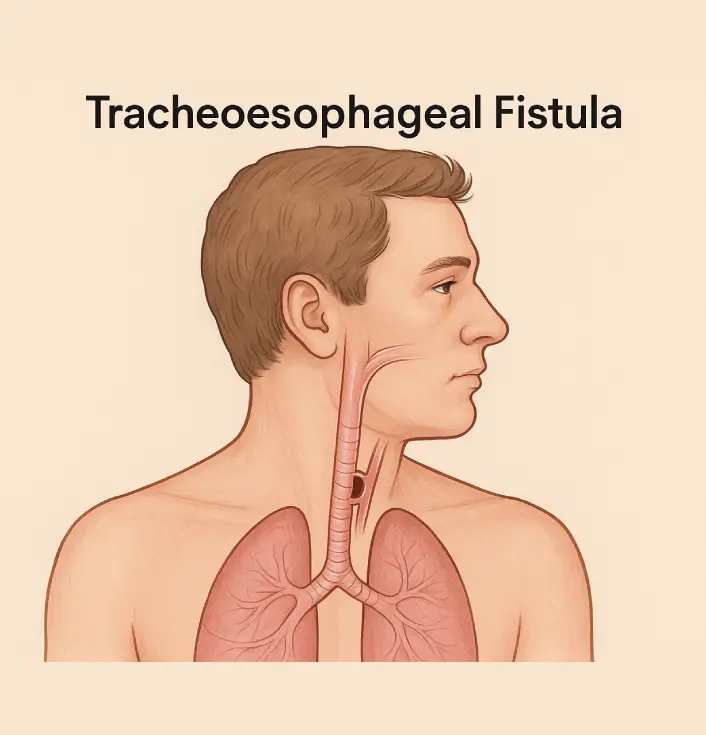
Definition:
Tracheoesophageal fistula (TEF) is a congenital abnormality characterized by an abnormal connection (fistula) between the trachea (windpipe) and the esophagus (food pipe). This condition leads to difficulty in swallowing, choking, and aspiration of food or fluids into the lungs, leading to respiratory distress.
Etiology (Causes):
The exact cause of TEF is not well understood, but it is believed to result from defective embryonic development of the trachea and esophagus during the 4th to 6th week of gestation. Possible risk factors include:
- Genetic mutations
- Environmental factors (e.g., maternal smoking, alcohol consumption)
- Associated with chromosomal abnormalities such as VACTERL syndrome (Vertebral defects, Anal atresia, Cardiac defects, Tracheoesophageal fistula, Esophageal atresia, Renal abnormalities, Limb abnormalities)
- Prematurity
Pathophysiology:
- Normally, the trachea and esophagus develop from the primitive foregut and separate by the tracheoesophageal septum.
- In TEF, the separation is incomplete, resulting in a connection between the two structures.
- Different types of TEF exist, with Type C (proximal esophageal atresia with distal TEF) being the most common (85% cases).
- The abnormal connection allows gastric contents to reflux into the trachea and lungs, leading to aspiration pneumonia, respiratory distress, and nutritional deficiencies.
Types of TEF:
- Type A – Esophageal atresia without fistula (10%)
- Type B – Proximal esophageal fistula with distal atresia (very rare)
- Type C – Proximal esophageal atresia with distal fistula (85% of cases)
- Type D – Proximal and distal fistulas with esophageal atresia (rare)
- Type E (H-type fistula) – TEF without esophageal atresia (4%)
Clinical Manifestations (Signs & Symptoms):
- Classic symptoms (3 Cs):
- Coughing
- Choking
- Cyanosis
- Excessive drooling and salivation
- Frothy bubbles in the mouth and nose
- Difficulty swallowing
- Respiratory distress, especially after feeding
- Aspiration pneumonia
- Abdominal distension (due to air entering the stomach through the fistula)
Diagnosis:
- Prenatal Diagnosis:
- Ultrasound: Polyhydramnios (excess amniotic fluid) is a suggestive finding.
- Postnatal Diagnosis:
- Clinical examination: Difficulty feeding, respiratory distress.
- Failure of Nasogastric Tube Insertion: An X-ray with contrast confirms the tube coiled in the upper pouch.
- Barium Swallow or Contrast Study: Detects fistula.
- Bronchoscopy or Esophagoscopy: Identifies the fistula directly.
- Echocardiogram & Renal Ultrasound: To rule out VACTERL-associated anomalies.
Medical Management:
- Preoperative Care:
- Maintain airway patency: Position baby at 30-45° upright
- Nasogastric Tube (Replogle tube) to suction saliva and prevent aspiration.
- IV Fluids and Parenteral Nutrition: Prevent dehydration and malnutrition.
- Oxygen Therapy: If needed for respiratory distress.
- Broad-Spectrum Antibiotics: To prevent pneumonia.
- Acid Suppressants (H2 blockers or PPI): Reduce gastric reflux and aspiration risk.
Surgical Management:
- Primary Repair (preferred in stable newborns)
- Closing the fistula and reconnecting the esophagus if there is a gap.
- Done within the first 24-48 hours of life in stable infants.
- Staged Repair (for high-risk infants or long-gap atresia)
- Temporary gastrostomy tube (G-tube) placement for feeding.
- Delayed esophageal repair after 2-3 months.
- Postoperative Care:
- Endotracheal intubation and ventilation if respiratory distress is severe.
- Chest tube placement if a thoracotomy is performed.
- Monitor for complications such as anastomotic leaks, strictures, GERD, or recurrent fistula.
Nursing Management:
Preoperative Nursing Care:
- Assess & Monitor:
- Respiratory distress, cyanosis, excessive drooling.
- Monitor oxygen saturation & arterial blood gases (ABGs).
- Maintain Airway & Prevent Aspiration:
- Positioning: Baby should be placed semi-upright (30-45°) with head elevated.
- Suctioning: Frequent oropharyngeal suctioning to prevent choking.
- Avoid oral feeds; administer IV fluids and parenteral nutrition.
- Educate Parents: Explain the need for surgery, care, and long-term prognosis.
Postoperative Nursing Care:
- Airway Management:
- Monitor for respiratory distress.
- Administer oxygen if needed.
- Positioning to prevent reflux and aspiration.
- Gastrostomy Care (if placed):
- Proper tube feeding technique.
- Monitor for infection at the insertion site.
- Pain Management:
- Administer analgesics as prescribed.
- Monitor for signs of pain (irritability, crying, increased heart rate).
- Monitor for Complications:
- Anastomotic leaks: Signs include tachycardia, increased respiratory rate, and fever.
- Esophageal stricture: Assess for difficulty swallowing, choking, or vomiting.
- Gastroesophageal reflux disease (GERD): Give proton pump inhibitors (PPIs) if needed.
- Recurrent fistula formation: Observe for choking and respiratory symptoms.
- Parental Education:
- Teach feeding techniques (e.g., small, slow feeds post-recovery).
- Educate about signs of aspiration pneumonia (coughing, difficulty breathing, fever).
- Encourage follow-up visits for esophageal dilation if strictures develop.
Complications of TEF:
- Aspiration Pneumonia
- Failure to Thrive (due to feeding difficulties)
- Tracheomalacia (softening of tracheal cartilage causing airway collapse)
- Anastomotic Stricture (narrowing of esophagus at the surgical site)
- Gastroesophageal Reflux Disease (GERD)
Prognosis:
- With early surgical intervention, the survival rate is over 90%.
- Long-term follow-up is needed for issues such as feeding difficulties, GERD, and respiratory infections.
Diaphragmatic Hernia (Congenital Diaphragmatic Hernia – CDH)
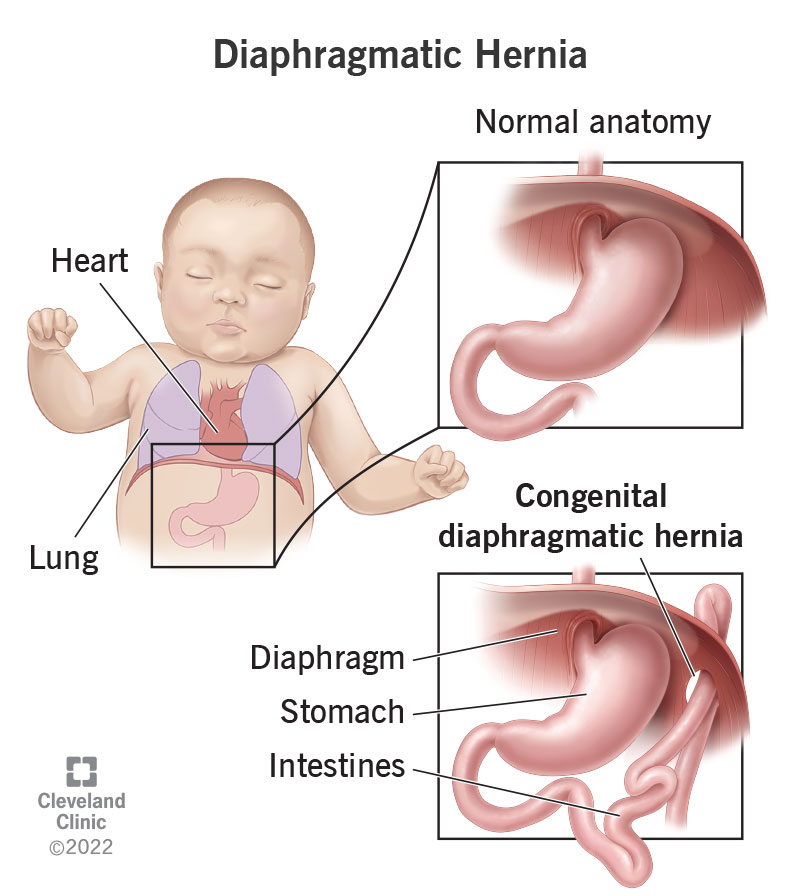
Definition:
Diaphragmatic hernia (CDH) is a congenital anomaly characterized by an abnormal opening in the diaphragm, allowing abdominal organs (stomach, intestines, liver, spleen) to move into the thoracic cavity. This leads to lung compression, pulmonary hypoplasia, and respiratory distress after birth.
Etiology (Causes):
The exact cause of CDH is unknown, but it is believed to result from genetic and environmental factors affecting fetal development during the 8th-10th week of gestation.
Risk Factors:
- Genetic mutations (e.g., Trisomy 18, Trisomy 21)
- Chromosomal anomalies (associated with 10-20% of cases)
- Vitamin A deficiency during pregnancy
- Maternal exposure to teratogens (drugs, alcohol, smoking)
- Congenital Syndromes:
- Fryns syndrome
- Cornelia de Lange syndrome
- Beckwith-Wiedemann syndrome
Types of CDH:
- Bochdalek Hernia (90%):
- Posterolateral defect, usually on the left side.
- Most common and severe type.
- Morgagni Hernia (5-10%):
- Anterior defect in the diaphragm.
- Usually asymptomatic and diagnosed later in life.
- Hiatal Hernia:
- Esophageal hiatus enlargement, allowing stomach protrusion into the thorax.
Pathophysiology:
- Failure of the diaphragm to fully develop allows abdominal contents to herniate into the thoracic cavity.
- Compression of developing lungs leads to pulmonary hypoplasia (underdeveloped lungs).
- Compression of heart & great vessels causes decreased cardiac output and hypoxia.
- Pulmonary Hypertension: Blood vessels in the underdeveloped lungs become thicker and constricted, leading to persistent pulmonary hypertension of the newborn (PPHN).
- Respiratory distress occurs immediately after birth due to poor lung expansion and hypoxia.
Clinical Manifestations (Signs & Symptoms):
At Birth (Severe Cases)
- Severe respiratory distress (cyanosis, tachypnea, nasal flaring, grunting).
- Scaphoid (sunken) abdomen due to displacement of abdominal organs.
- Barrel-shaped chest from compressed lung.
- Absence of breath sounds on the affected side.
- Heart sounds displaced to the right due to mediastinal shift.
- Severe hypoxia (low oxygen levels).
- Pulmonary hypertension leading to poor perfusion.
Later Childhood (Mild Cases)
- Frequent respiratory infections.
- Gastroesophageal reflux (GERD).
- Failure to thrive (poor weight gain).
- Recurrent abdominal pain.
Diagnosis:
Prenatal Diagnosis (Fetal Screening)
- Ultrasound (18-24 weeks gestation)
- Stomach and intestines visible in the chest.
- Small lung size (lung-to-head ratio – LHR).
- Fetal MRI or Fetal Echocardiography: To assess lung hypoplasia and heart displacement.
Postnatal Diagnosis
- Chest X-ray:
- Bowel loops, stomach, or liver visible in the thoracic cavity.
- Mediastinal shift (heart displaced).
- Ultrasound Abdomen & Chest: Confirms organ displacement.
- Blood Gas Analysis (ABG): Shows respiratory acidosis and hypoxia.
- Echocardiography: Assesses pulmonary hypertension.
Medical Management:
Preoperative Care (Before Surgery)
- Immediate respiratory support: Intubation with mechanical ventilation to prevent lung collapse.
- Avoid Bag-Mask Ventilation: It can push air into the stomach and worsen lung compression.
- Sedation & Paralysis: To prevent stress and hypoxia.
- Nitric Oxide Therapy: For persistent pulmonary hypertension.
- Extracorporeal Membrane Oxygenation (ECMO): If severe hypoxia persists.
Surgical Management:
Timing:
- Surgery is done once the baby’s condition is stabilized (usually within 48-72 hours).
Procedure:
- Primary Repair:
- The surgeon places the herniated abdominal organs back into the abdominal cavity.
- The diaphragmatic defect is closed with sutures or a synthetic patch.
- Patch Repair:
- For large defects, an artificial patch (e.g., Gore-Tex) is used to reinforce the diaphragm.
Postoperative Care:
- Mechanical ventilation & oxygen therapy until lung function improves.
- Fluid & electrolyte management.
- Pain control with opioids (Morphine, Fentanyl).
- Monitor for complications:
- Pulmonary hypertension
- Gastroesophageal reflux (GERD)
- Lung infections
- Bowel obstruction due to adhesion formation.
Nursing Management:
Preoperative Nursing Care:
Airway & Respiratory Management:
- Ensure Intubation: Avoid manual bag ventilation.
- Maintain mechanical ventilation with high-frequency oscillatory ventilation (HFOV).
- Positioning: Infant in semi-Fowler’s position (head elevated) to reduce abdominal pressure on lungs.
- Oxygen Therapy: Nitric oxide to reduce pulmonary hypertension.
Hemodynamic Stability:
- Monitor blood pressure for shock or low perfusion.
- IV Fluids to maintain hydration & electrolyte balance.
- Monitor Arterial Blood Gases (ABGs) for respiratory status.
Prevent Infection & Aspiration:
- Maintain strict asepsis in ventilated infants.
- Frequent oral and nasal suctioning.
- Administer antibiotics if infection is suspected.
Postoperative Nursing Care:
Respiratory Support:
- Continue ventilatory support.
- Weaning off the ventilator should be done gradually.
- Monitor oxygen saturation & ABGs to detect early signs of hypoxia.
Pain Management:
- Administer opioids (Morphine, Fentanyl) as prescribed.
- Monitor for signs of pain (tachycardia, irritability, high blood pressure).
Gastrointestinal & Nutrition Support:
- Gastrostomy feeding may be required initially.
- Gradual oral feeding when bowel function returns.
- Monitor for GERD & provide anti-reflux positioning.
Monitor for Complications:
- Pulmonary Hypertension – Monitor for cyanosis, tachycardia.
- GERD – Administer proton pump inhibitors (PPIs) if needed.
- Bowel Obstruction – Observe for abdominal distension, vomiting, or feeding intolerance.
Complications of CDH:
- Respiratory Failure due to lung hypoplasia.
- Persistent Pulmonary Hypertension (PPHN).
- Gastroesophageal Reflux Disease (GERD).
- Bowel obstruction due to adhesions.
- Neurodevelopmental delays (in severe cases).
Prognosis:
- Survival Rate:
- Mild Cases: 80-90% survival.
- Severe Cases with Pulmonary Hypertension: 50-70%.
- Long-term Issues:
- Chronic lung disease (oxygen dependency).
- GERD and feeding difficulties.
- Developmental delays (motor and cognitive).
Key Points:
✔ CDH is a life-threatening condition requiring emergency intervention.
✔ Early diagnosis (prenatal screening) improves survival.
✔ Surgical repair is essential but does not correct lung hypoplasia.
✔ Postoperative care focuses on lung recovery, oxygenation, and feeding.
Acute Nasopharyngitis (Common Cold)

Definition:
Acute nasopharyngitis, commonly known as the common cold, is a self-limiting viral infection of the upper respiratory tract affecting the nose and throat (nasopharynx). It is one of the most common childhood illnesses, characterized by nasal congestion, rhinorrhea, sore throat, cough, and mild fever.
Etiology (Causes):
Acute nasopharyngitis is primarily caused by viral infections, with over 200 different viruses identified. The most common pathogens include:
- Rhinovirus (most common, accounting for 30-50% of cases)
- Coronavirus
- Adenovirus
- Respiratory Syncytial Virus (RSV)
- Influenza and Parainfluenza viruses
- Enterovirus (e.g., Coxsackie virus)
Risk Factors:
- Age: Infants and young children are more prone due to immature immune systems.
- Season: More common in winter and rainy seasons.
- Exposure: Close contact in schools, daycare centers, and crowded areas.
- Weakened immunity: Malnutrition, underlying diseases.
- Allergies and Environmental factors: Exposure to dust, smoke, and pollution.
Pathophysiology:
- Viral Entry: The virus enters through inhalation or contact with contaminated surfaces.
- Attachment & Replication: The virus attaches to the nasal mucosa and begins replicating.
- Inflammatory Response:
- Causes edema, congestion, and mucus production in the nasal passages.
- Release of histamines and cytokines, leading to sore throat and nasal obstruction.
- Immune Response Activation:
- Fever develops as a systemic inflammatory response.
- Increased mucus production to trap and eliminate pathogens.
- Coughing and sneezing help to expel the virus.
- Resolution:
- In most cases, the immune system clears the virus within 7-10 days.
- Occasionally, secondary bacterial infections like sinusitis or otitis media can develop.
Clinical Manifestations (Signs & Symptoms):
Early Symptoms (1-3 days after exposure)
- Runny nose (rhinorrhea)
- Sneezing
- Sore throat
- Low-grade fever (usually below 101°F or 38.5°C)
- Mild headache
- Chills and malaise
Established Symptoms (3-5 days)
- Nasal congestion (stuffy nose)
- Cough (dry initially, may become productive)
- Hoarseness of voice
- Watery or red eyes
- Difficulty in breathing (especially in infants)
Late Symptoms (5-10 days)
- Thick, yellow or green nasal discharge (indicates mucus clearance, not necessarily bacterial infection)
- Persistent cough
- Fatigue
- Loss of appetite (especially in children)
Complications (if untreated or severe)
- Otitis media (middle ear infection)
- Sinusitis
- Bronchitis
- Pneumonia (rare but possible)
- Exacerbation of asthma in predisposed children
Diagnosis:
- Clinical Examination:
- Based on history and symptoms (nasal congestion, cough, sore throat).
- Inspection of throat (redness), nasal mucosa (swelling), and lymph nodes (enlarged).
- Laboratory Tests (if needed for complications):
- Throat Swab: For Group A Streptococcus if bacterial pharyngitis is suspected.
- Nasopharyngeal Swab: For Influenza, RSV, or other viral panels.
- Complete Blood Count (CBC):
- Viral infection: Normal or slightly increased lymphocyte count.
- Bacterial infection: Elevated neutrophils.
- Radiological Tests (If Complications Suspected)
- Chest X-ray: If pneumonia is suspected.
- Sinus X-ray or CT scan: If sinusitis is suspected.
Medical Management:
There is no specific antiviral treatment for acute nasopharyngitis; supportive care is the mainstay of treatment.
1. Symptomatic Treatment:
- Rest & Hydration: Encouraging fluids helps thin mucus and prevents dehydration.
- Paracetamol (Acetaminophen): For fever and body aches.
- Ibuprofen: For fever and pain relief (avoid in infants <6 months).
- Saline Nasal Drops: Helps relieve nasal congestion.
- Steam Inhalation: Soothes the throat and loosens mucus.
- Honey (for children >1 year): Natural cough suppressant.
2. Medications (as needed):
- Decongestants (e.g., Xylometazoline, Phenylephrine nasal drops) – Only for short-term use (3-5 days).
- Antihistamines (e.g., Cetirizine, Loratadine) – For allergic rhinitis component.
- Cough Suppressants (e.g., Dextromethorphan) – Used for severe dry cough.
- Expectorants (e.g., Guaifenesin) – Helps loosen mucus.
3. Antibiotics (ONLY if bacterial complications arise)
- If secondary bacterial infection (e.g., sinusitis, otitis media, pneumonia) is confirmed, antibiotics like Amoxicillin-Clavulanate or Azithromycin may be prescribed.
Surgical Management:
Surgical intervention is not required for acute nasopharyngitis unless there are complications such as:
- Adenoidectomy or Tonsillectomy: If there are recurrent throat infections or chronic nasal obstruction due to enlarged adenoids.
- Sinus Drainage Surgery: For chronic sinusitis due to persistent bacterial infection.
- Ear Tube Placement: If recurrent otitis media occurs due to persistent fluid in the middle ear.
Nursing Management:
1. Assessment:
- Monitor respiratory status (breathing pattern, wheezing, nasal flaring).
- Assess hydration status (urine output, mucous membrane dryness).
- Check for fever and other systemic symptoms.
- Observe for complications like ear pain (otitis media), persistent cough (bronchitis), or high fever (secondary bacterial infection).
2. Nursing Interventions:
A. Airway Clearance:
- Encourage nasal suctioning in infants (use bulb syringe).
- Elevate the child’s head to ease breathing.
- Encourage deep breathing and coughing in older children.
- Provide humidified air or steam inhalation.
B. Fever Management:
- Administer antipyretics (Paracetamol, Ibuprofen) as prescribed.
- Encourage tepid sponge bath if fever is high.
- Monitor temperature regularly.
C. Hydration & Nutrition:
- Encourage increased fluid intake (warm water, soups, fruit juices).
- Administer oral rehydration solutions (ORS) if mild dehydration is present.
- Small frequent meals to maintain nutrition.
D. Comfort Measures:
- Use warm saline gargles for sore throat.
- Encourage rest to aid recovery.
- Advise parents to avoid giving aspirin due to the risk of Reye’s syndrome.
3. Patient and Parent Education:
- Hand Hygiene: Frequent handwashing prevents transmission.
- Avoid crowded places to reduce spread.
- Teach cough etiquette (covering mouth, disposing of tissues properly).
- Avoid overuse of antibiotics as it does not treat viral infections.
Prognosis:
- Acute nasopharyngitis resolves within 7-10 days with proper care.
- If managed well, complications are rare.
- Recurrent colds are common in young children, but as immunity develops, the frequency decreases.
Tonsillitis in Children
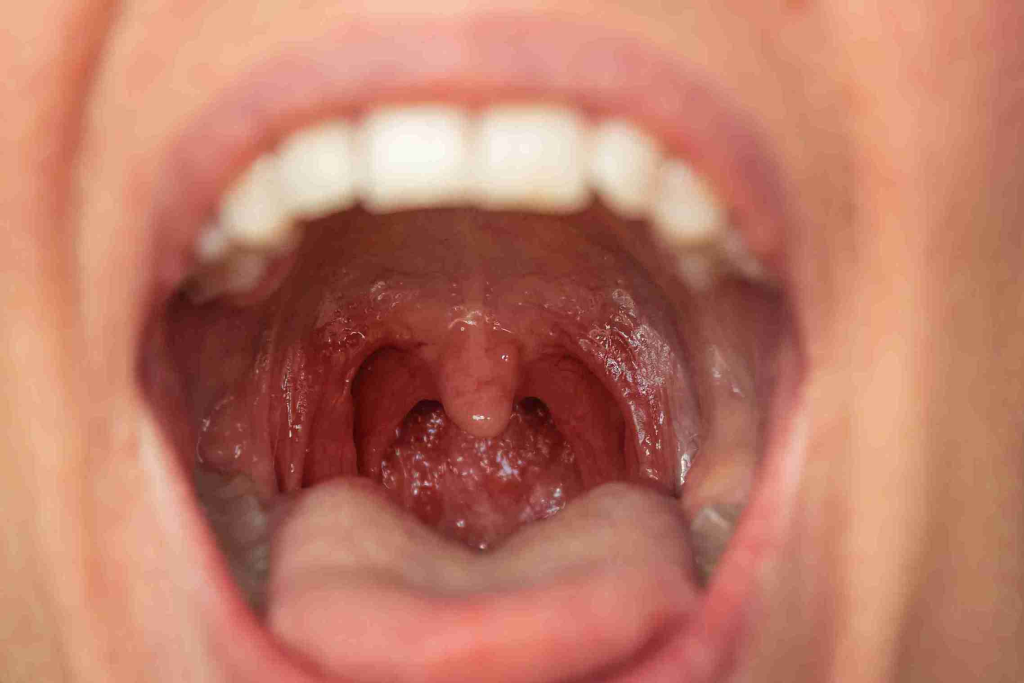
Definition:
Tonsillitis is an inflammation of the tonsils, two oval-shaped lymphoid tissues located at the back of the throat. It is commonly caused by viral or bacterial infections, leading to symptoms such as sore throat, difficulty swallowing, fever, and swollen lymph nodes.
Etiology (Causes):
Tonsillitis can be viral or bacterial in origin.
1. Viral Causes (Most Common – 70-80%):
- Adenovirus (most common)
- Rhinovirus
- Influenza virus
- Parainfluenza virus
- Epstein-Barr virus (EBV) – Causes infectious mononucleosis.
- Herpes Simplex Virus (HSV)
2. Bacterial Causes (20-30%):
- Group A Beta-Hemolytic Streptococcus (GABHS) → Causes Streptococcal Tonsillitis (Strep Throat).
- Streptococcus pneumoniae
- Haemophilus influenzae
- Staphylococcus aureus
- Moraxella catarrhalis
Risk Factors:
- Age: Common in children 3-15 years.
- Frequent upper respiratory infections.
- Exposure to infected individuals.
- Poor hygiene and crowded places.
Pathophysiology:
- Pathogen Entry: The virus or bacteria enters through airborne droplets or direct contact.
- Invasion of Tonsillar Tissue: The pathogen multiplies in the crypts of the tonsils, causing inflammation and swelling.
- Immune Response Activation:
- The body releases white blood cells to fight infection.
- Pus formation in bacterial tonsillitis.
- Swelling of tonsils and throat leads to pain and difficulty swallowing.
- Secondary Complications (if untreated):
- Abscess formation (Peritonsillar Abscess)
- Rheumatic fever (due to untreated streptococcal infection).
- Post-streptococcal glomerulonephritis.
Clinical Manifestations (Signs & Symptoms):
General Symptoms:
- Sore throat (persistent or severe)
- Painful swallowing (Dysphagia)
- Fever (high-grade in bacterial infections)
- Malaise, headache, and body aches
- Bad breath (halitosis)
- Voice changes (muffled voice or “hot potato voice”)
- Swollen lymph nodes (especially in bacterial tonsillitis)
Physical Examination Findings:
- Enlarged, red tonsils.
- White or yellow exudates (pus) on tonsils (seen in bacterial infections).
- Palatal petechiae (red spots on the soft palate).
- Tender cervical lymph nodes.
- Strawberry tongue (seen in Streptococcal infection).
- Drooling (if severe pain prevents swallowing).
Complications (If Untreated):
- Peritonsillar Abscess: Pus collection around the tonsils.
- Airway Obstruction: Due to severe swelling.
- Otitis Media (Middle Ear Infection): Infection spreading to the ear.
- Rheumatic Fever: Due to untreated GABHS infection.
- Post-Streptococcal Glomerulonephritis (PSGN): Kidney inflammation after streptococcal infection.
Diagnosis:
1. Clinical Examination:
- Throat Examination: Red, swollen tonsils, pus on tonsils, enlarged lymph nodes.
- Fever and Pain Assessment.
2. Laboratory Tests:
- Throat Swab Culture: Identifies Group A Streptococcus (GABHS).
- Rapid Antigen Detection Test (RADT): Quick test for streptococcal infection.
- Complete Blood Count (CBC):
- Viral Infection: Increased lymphocytes.
- Bacterial Infection: Increased neutrophils and high WBC count.
- Anti-Streptolysin O (ASO) Titer: Confirms recent streptococcal infection.
3. Imaging (If Needed):
- X-ray or CT scan: If abscess formation or airway obstruction is suspected.
Medical Management:
1. Symptomatic Treatment (For Viral Tonsillitis)
- Supportive care is the mainstay (No antibiotics needed).
- Rest & Fluids: Encourages healing.
- Pain Relief:
- Paracetamol (Acetaminophen) or Ibuprofen.
- Gargles: Warm saline gargles help reduce throat pain.
- Throat Lozenges: Provide temporary pain relief.
- Humidified Air: Reduces throat irritation.
- Soft Diet & Cold Fluids: Helps ease swallowing.
2. Antibiotic Therapy (For Bacterial Tonsillitis)
- First-line: Penicillin V or Amoxicillin (10 days course).
- Alternative: Azithromycin or Cephalosporins (if allergic to penicillin).
- IV Antibiotics: Needed in severe cases or if complications arise.
Surgical Management:
1. Tonsillectomy (Removal of Tonsils)
Indications:
- Recurrent Tonsillitis (≥7 episodes/year or ≥5 episodes/year for 2 consecutive years).
- Chronic Tonsillitis causing difficulty in eating and speaking.
- Peritonsillar Abscess (if unresponsive to antibiotics).
- Obstructive Sleep Apnea (OSA)
- Airway obstruction due to enlarged tonsils.
Procedure:
- Done under general anesthesia.
- Tonsils are removed using scalpel, electrocautery, or laser.
- Outpatient procedure with a 1-2 week recovery period.
Post-Tonsillectomy Care:
- Soft, cold diet (ice cream, yogurt, soups).
- Avoid hard/spicy foods to prevent irritation.
- Adequate hydration.
- Pain management (Paracetamol or Ibuprofen).
- Monitor for bleeding (rare but serious complication).
Nursing Management:
Preoperative Nursing Care (If Surgery is Planned):
- Assess for Infection: Ensure no active throat infection before surgery.
- Educate Parents & Child:
- Explain procedure and recovery.
- Advise on pain management post-surgery.
- Fasting Before Surgery:
- NPO (nothing by mouth) for 6-8 hours before surgery.
Postoperative Nursing Care (After Tonsillectomy):
Airway & Breathing:
- Positioning: Child should be placed in side-lying or semi-Fowler’s position.
- Suctioning: Gentle suctioning if needed (avoid trauma).
Pain Management:
- Administer prescribed analgesics (Paracetamol/Ibuprofen).
- Ice packs on the throat to reduce swelling.
- Encourage cold fluids & ice pops.
Monitor for Bleeding (First 24 Hours & 7-10 Days Later):
- Bright red bleeding from throat → Emergency!
- Frequent swallowing → May indicate bleeding.
- Pale skin, dizziness, tachycardia → Sign of blood loss.
Dietary Guidelines:
- Start with clear liquids (water, juice, ice chips).
- Soft foods (mashed potatoes, yogurt) after 24 hours.
- Avoid acidic, spicy, and crunchy foods for 2 weeks.
Activity Restrictions:
- Avoid strenuous activities for 2 weeks.
- No heavy lifting or vigorous exercise.
Parental Education:
- Signs of Infection or Bleeding → Seek immediate medical attention.
- Encourage proper oral hygiene (gentle mouth rinsing, no brushing near the surgical site).
Prognosis:
- Viral tonsillitis: Resolves within 7-10 days.
- Bacterial tonsillitis: Improves within 48 hours of antibiotics.
- Tonsillectomy: Provides permanent relief for recurrent cases.
Croup (Laryngotracheobronchitis)
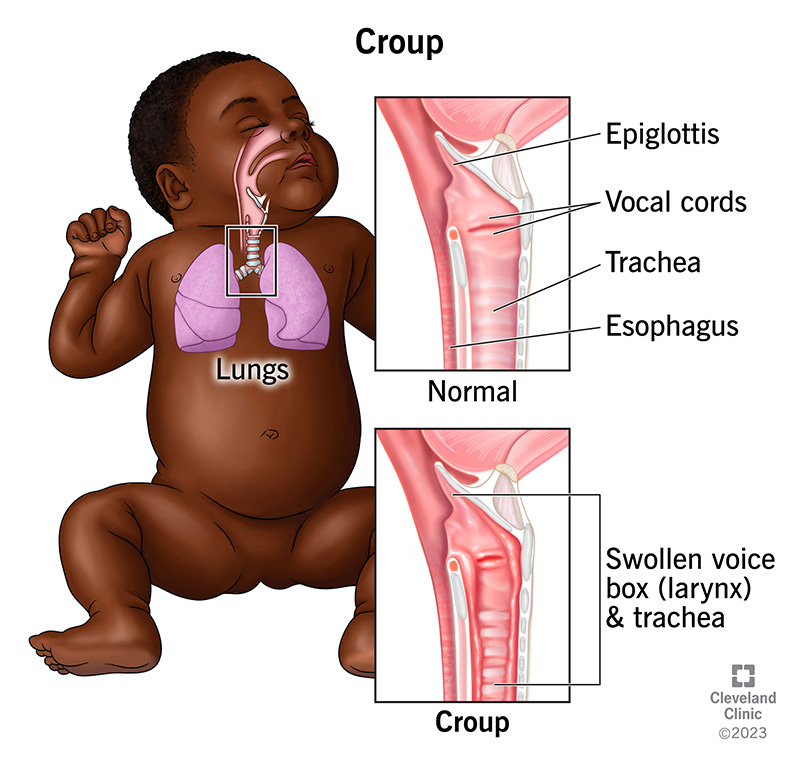
Definition:
Croup is a common upper respiratory tract infection in children, characterized by inflammation and swelling of the larynx (voice box), trachea (windpipe), and bronchi. It leads to a barking cough, stridor (high-pitched breathing sound), and respiratory distress.
Etiology (Causes):
Croup is primarily caused by viral infections affecting the upper airway. The most common viruses include:
1. Viral Causes (Most Common – 75-85%)
- Parainfluenza virus (Types 1, 2, and 3) – Most common
- Respiratory Syncytial Virus (RSV)
- Influenza A and B
- Adenovirus
- Measles virus
- Coronavirus
- Enteroviruses
2. Bacterial Causes (Rare – Can Cause Severe Croup)
- Staphylococcus aureus
- Streptococcus pneumoniae
- Haemophilus influenzae
- Moraxella catarrhalis
Risk Factors:
- Age: Most common in children 6 months to 3 years.
- Season: More common in fall and winter.
- Previous respiratory infections.
- Exposure to sick individuals.
- Allergies and airway sensitivity.
Pathophysiology:
- Viral infection of the upper airway triggers inflammation.
- Edema and swelling of the larynx, trachea, and bronchi lead to airway narrowing.
- Increased mucus production further obstructs airflow.
- Stridor (high-pitched sound during breathing) occurs due to turbulent airflow in the narrowed airway.
- Inspiratory and expiratory distress due to increased work of breathing.
- Severe cases: Progressive airway obstruction can lead to hypoxia and respiratory failure.
Clinical Manifestations (Signs & Symptoms):
Croup symptoms usually begin gradually over 24-48 hours and worsen at night.
Early Symptoms:
- Mild fever (38-39°C or 100.4-102.2°F)
- Runny nose, nasal congestion
- Hoarseness
- Sore throat
Established Symptoms:
- Characteristic “Barking” Cough (resembles a seal’s bark).
- Inspiratory Stridor (high-pitched noise during breathing).
- Respiratory distress (retractions, nasal flaring, tachypnea).
- Restlessness, irritability, or anxiety.
- Worse symptoms at night.
Severe Symptoms (Severe Croup):
- Stridor at rest (not just during crying).
- Severe retractions (chest pulling in with breathing).
- Cyanosis (blue skin, lips, or fingertips) – Indicates hypoxia.
- Lethargy, difficulty in breathing (ominous sign of respiratory failure).
Diagnosis:
1. Clinical Examination:
- History of barking cough, stridor, hoarseness, and night-time worsening symptoms.
- Assess respiratory distress: Retractions, nasal flaring, accessory muscle use.
2. Radiological Tests (If Needed for Severe Cases):
- Neck X-ray (Soft Tissue X-ray)
- Steeple Sign: Narrowing of the subglottic region of the trachea (seen in croup).
3. Laboratory Tests:
- Pulse Oximetry: To check oxygen saturation.
- Throat Swab or Viral PCR: Identifies the causative virus.
- Arterial Blood Gas (ABG) Analysis: Assesses oxygen levels in severe cases.
Medical Management:
1. Mild Croup (No Stridor at Rest)
- Supportive Care:
- Humidified air (cool mist, steamy bathroom).
- Hydration (fluids to prevent dehydration).
- Rest and comfort measures.
- Medications:
- Oral Dexamethasone (0.6 mg/kg, single dose) – Reduces airway swelling.
- Paracetamol or Ibuprofen for fever and discomfort.
2. Moderate Croup (Stridor at Rest, Mild Respiratory Distress)
- Nebulized Epinephrine (2.25% racemic epinephrine) – Provides rapid relief by reducing airway swelling.
- Oral or IV Corticosteroids (Dexamethasone/Prednisolone).
- Oxygen Therapy (If O2 saturation <92%).
3. Severe Croup (Severe Respiratory Distress, Cyanosis)
- Hospitalization required.
- High-dose nebulized epinephrine for immediate relief.
- Intravenous (IV) corticosteroids (Dexamethasone 0.6 mg/kg).
- Continuous oxygen therapy.
- Endotracheal Intubation (If Airway Obstruction Progresses).
Surgical Management:
Rarely Required Unless Severe Complications Arise
- Tracheostomy – If airway obstruction is life-threatening and non-responsive to medical therapy.
- Endotracheal Intubation – For severe respiratory failure.
Nursing Management:
1. Assessment:
- Monitor Respiratory Status:
- Assess rate, depth, and effort of breathing.
- Monitor for stridor, nasal flaring, retractions.
- Check oxygen saturation (SpO2).
- Monitor for Signs of Hypoxia:
- Cyanosis, restlessness, confusion.
- Assess Hydration Status:
- Monitor urine output, skin turgor, mucous membrane dryness.
2. Nursing Interventions:
A. Airway and Breathing Management
- Positioning: Keep child in semi-Fowler’s or upright position to ease breathing.
- Humidified Oxygen: Helps moisten airways and reduce inflammation.
- Administer Nebulized Epinephrine: To rapidly reduce airway swelling.
- Give Corticosteroids (Dexamethasone) as prescribed.
- Monitor for Stridor and Retractions.
B. Comfort and Hydration
- Encourage Fluids to prevent dehydration.
- Use cool-mist humidifiers or take the child outside for cool air relief.
- Encourage rest and minimal crying (crying worsens airway obstruction).
- Administer fever reducers (Paracetamol/Ibuprofen).
C. Infection Control
- Isolate child if viral croup is suspected to prevent spreading.
- Teach hand hygiene and respiratory hygiene to caregivers.
3. Parent and Child Education:
- Encourage Home Care for Mild Cases:
- Increase fluid intake.
- Use humidified air (cool mist therapy).
- Administer prescribed steroids if needed.
- When to Seek Emergency Care:
- Persistent stridor at rest.
- Severe difficulty in breathing (chest retractions, nasal flaring).
- Bluish lips or skin (cyanosis).
- Lethargy or extreme agitation.
- Avoid Over-The-Counter Cough Syrups: Can suppress necessary cough reflex.
Complications of Croup:
- Severe Airway Obstruction → Respiratory Failure.
- Bacterial Superinfection (Bacterial Tracheitis, Pneumonia).
- Dehydration due to poor oral intake.
- Ear Infections (Otitis Media).
- Rare: Myocarditis or Neurological Complications (encephalitis).
Prognosis:
- Mild to moderate cases recover within 3-7 days.
- Severe cases may require hospitalization but usually recover with treatment.
- Recurrences can happen, but severity often decreases with age.
Key Points:
✔ Croup is a common viral infection in young children causing airway inflammation.
✔ Barking cough, stridor, and respiratory distress are characteristic symptoms.
✔ Mild cases can be managed at home; severe cases need nebulized epinephrine & steroids.
✔ Nursing care focuses on airway management, hydration, and education.
✔ Prompt intervention prevents complications like respiratory failure.
Bronchitis in Children
Definition:
Bronchitis is the inflammation of the bronchi (large airways) in the lungs, leading to increased mucus production, cough, and airway obstruction. It can be acute (short-term, lasting less than 3 weeks) or chronic (lasting more than 3 months per year for two consecutive years).
Etiology (Causes):
1. Acute Bronchitis (Most Common in Children)
Caused by viral infections affecting the respiratory tract.
- Viral Causes (Most Common – 90%):
- Influenza A & B
- Respiratory Syncytial Virus (RSV)
- Adenovirus
- Parainfluenza virus
- Coronavirus
- Rhinovirus (Common Cold Virus)
- Bacterial Causes (Less Common – 10%):
- Mycoplasma pneumoniae
- Chlamydia pneumoniae
- Streptococcus pneumoniae
- Bordetella pertussis (Whooping Cough)
- Other Causes:
- Exposure to pollutants (smoke, dust, fumes)
- Allergic reactions (asthmatic bronchitis)
- Gastroesophageal reflux disease (GERD)
- Post-nasal drip (sinus infections leading to cough)
2. Chronic Bronchitis
- Most commonly seen in children with underlying lung diseases like cystic fibrosis, asthma, or chronic exposure to irritants (e.g., passive smoking).
Pathophysiology:
- Irritants (viruses, bacteria, pollutants) cause inflammation of the bronchial mucosa.
- Inflammation leads to increased mucus production, narrowing the airway.
- Ciliary dysfunction (impaired mucus clearance) results in accumulation of mucus.
- Bronchial hyperreactivity leads to persistent cough and airway obstruction.
- In severe cases, airway remodeling may occur, leading to chronic bronchitis.
Clinical Manifestations (Signs & Symptoms):
Acute Bronchitis
- Persistent dry or productive cough (lasting 1-3 weeks)
- Wheezing or mild shortness of breath
- Low-grade fever (may be absent)
- Sore throat, runny nose (if viral infection present)
- Chest discomfort or pain due to excessive coughing
- Mild fatigue and malaise
- Clear, yellow, or greenish sputum (not always present in viral cases)
Severe Symptoms (Seek Emergency Care)
- High fever (>102°F or 39°C)
- Difficulty breathing (retractions, nasal flaring, cyanosis)
- Persistent wheezing or stridor
- Severe fatigue or lethargy
- Signs of dehydration (dry lips, low urine output, sunken eyes)
Chronic Bronchitis
- Cough lasting >3 months per year for 2 consecutive years
- Frequent respiratory infections
- Shortness of breath
- Excessive mucus production
- Wheezing (especially in children with asthma or allergies)
Diagnosis:
1. Clinical Examination:
- History of prolonged cough (1-3 weeks).
- Auscultation: Wheezing, crackles, or rhonchi (coarse breath sounds).
- Chest discomfort, signs of respiratory distress.
2. Laboratory Tests:
- Complete Blood Count (CBC):
- Elevated lymphocytes (viral infection)
- Elevated neutrophils (bacterial infection)
- Sputum Culture: Identifies bacterial infection if present.
- Nasopharyngeal Swab PCR Test: Detects viral pathogens (RSV, influenza, COVID-19).
3. Imaging:
- Chest X-ray (to rule out pneumonia):
- Usually normal in viral bronchitis.
- Shows bronchial thickening or infiltrates in bacterial infections.
- Pulmonary Function Test (Spirometry) (if chronic bronchitis suspected):
- Measures airway obstruction and lung capacity.
- Pulse Oximetry: Assesses oxygen levels, especially if respiratory distress is present.
Medical Management:
1. Supportive Treatment (For Viral Bronchitis)
- Rest and Hydration: Encourages mucus clearance.
- Antipyretics (Paracetamol/Ibuprofen): Reduces fever and discomfort.
- Humidified Air or Steam Inhalation: Loosens mucus and soothes airways.
- Cough Syrups:
- Dextromethorphan (for dry cough in older children).
- Expectorants (Guaifenesin): Helps loosen mucus.
2. Medications (If Needed)
- Bronchodilators (Salbutamol, Albuterol Nebulization)
- Used in children with wheezing or bronchospasm.
- Steroids (Oral Prednisolone or Inhaled Budesonide)
- Used if airway inflammation is severe.
- Antibiotics (ONLY if bacterial bronchitis is confirmed)
- Amoxicillin-Clavulanate or Azithromycin.
- For atypical bacteria (Mycoplasma pneumoniae) → Macrolides (Clarithromycin).
- Antiviral Drugs (If Influenza is Confirmed)
- Oseltamivir (Tamiflu) within 48 hours of symptoms.
Surgical Management:
Surgery is not required for bronchitis unless complications arise:
- Bronchoscopy (to remove mucus plugs in severe cases).
- Lung Surgery (for chronic bronchitis with severe lung damage, rare in children).
Nursing Management:
1. Assessment:
- Monitor respiratory status: Check for wheezing, tachypnea, retractions, cyanosis.
- Assess hydration status: Monitor urine output, skin turgor, mucous membranes.
- Check oxygen saturation (SpO2): Ensure oxygen levels >92%.
2. Nursing Interventions:
A. Airway & Breathing Management
- Positioning: Keep child in semi-Fowler’s or upright position to ease breathing.
- Oxygen Therapy: If SpO2 < 92% or respiratory distress is present.
- Nebulization with Salbutamol (if wheezing is present).
- Encourage Deep Breathing & Coughing to mobilize secretions.
B. Fever & Pain Management
- Administer Paracetamol/Ibuprofen for fever and discomfort.
- Provide warm fluids (soups, teas) to soothe the throat.
C. Hydration & Nutrition
- Encourage fluid intake to thin mucus.
- Offer small, frequent meals to maintain energy levels.
D. Infection Control
- Educate caregivers about proper hand hygiene.
- Encourage respiratory etiquette (covering mouth while coughing).
- Limit exposure to smoke, dust, and allergens.
3. Parent & Child Education:
- Teach Parents When to Seek Medical Help:
- Persistent high fever (>102°F).
- Difficulty in breathing (severe wheezing, cyanosis, lethargy).
- Cough lasting >3 weeks or worsening symptoms.
- Avoid unnecessary antibiotics for viral bronchitis.
- Importance of Flu & Pneumococcal Vaccination.
Complications of Bronchitis:
- Pneumonia (Bacterial Superinfection).
- Bronchiolitis (in infants <2 years).
- Chronic obstructive airway disease (rare but possible in severe chronic bronchitis).
- Respiratory Failure (in extreme cases).
Prognosis:
- Acute bronchitis usually resolves within 10-14 days with symptomatic treatment.
- Chronic bronchitis requires long-term management (especially in children with asthma or cystic fibrosis).
Key Points:
✔ Bronchitis is a common viral illness in children, causing persistent cough and mucus production.
✔ Most cases are viral and require supportive care, not antibiotics.
✔ Severe cases need bronchodilators, steroids, or hospitalization for oxygen therapy.
✔ Preventive measures include vaccination, hand hygiene, and avoiding pollutants.
Bronchiolitis in Children
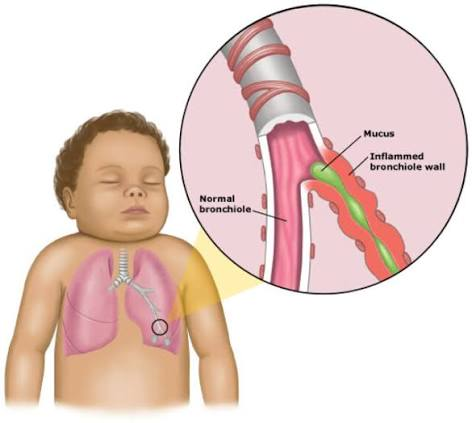
Definition:
Bronchiolitis is a common viral lower respiratory tract infection in infants and young children, characterized by inflammation and obstruction of the small airways (bronchioles). It leads to wheezing, difficulty breathing, cough, and respiratory distress.
Etiology (Causes):
Bronchiolitis is primarily caused by viral infections, with Respiratory Syncytial Virus (RSV) being the most common.
1. Viral Causes (Most Common – 90%)
- Respiratory Syncytial Virus (RSV) – 70-80% of cases
- Adenovirus
- Influenza virus (A & B)
- Parainfluenza virus
- Human metapneumovirus
- Rhinovirus
- Coronavirus
2. Risk Factors:
- Age: Most common in infants <2 years, peak at 2-6 months.
- Premature birth (<37 weeks gestation).
- Low birth weight.
- Congenital heart disease.
- Chronic lung diseases (e.g., cystic fibrosis, bronchopulmonary dysplasia).
- Exposure to tobacco smoke.
- Lack of breastfeeding.
- Crowded environments (daycare centers, siblings in school).
Pathophysiology:
- Virus infects epithelial cells of the bronchioles → causes inflammation & cell death.
- Edema, mucus production, and debris accumulation lead to airway obstruction.
- Narrowing of bronchioles → airflow limitation → wheezing & difficulty breathing.
- Air trapping & atelectasis (collapsed alveoli) cause hypoxia (low oxygen levels).
- Increased work of breathing → leads to respiratory distress & fatigue.
- Severe cases can lead to respiratory failure due to exhaustion & hypoxemia.
Clinical Manifestations (Signs & Symptoms):
Early Symptoms (1-3 Days)
- Runny nose (rhinorrhea)
- Mild fever (38-39°C or 100.4-102.2°F)
- Mild cough
- Decreased appetite, irritability
Progressive Symptoms (3-7 Days)
- Wheezing (high-pitched sound during breathing)
- Tachypnea (rapid breathing >60 breaths/min in infants)
- Nasal flaring, retractions (chest wall pulling in during breathing)
- Grunting, head bobbing (severe cases)
- Cyanosis (bluish skin, lips, or fingertips in severe hypoxia)
- Poor feeding & dehydration (due to increased breathing effort)
Severe Symptoms (Need Emergency Care)
- Severe respiratory distress (nasal flaring, severe retractions)
- Apnea (temporary pauses in breathing) in preterm infants
- Oxygen saturation <92%
- Lethargy or extreme irritability
- Central cyanosis (blue discoloration around lips & tongue)
Diagnosis:
1. Clinical Examination:
- History of recent upper respiratory infection
- Auscultation: Wheezing, crackles, prolonged expiratory phase.
- Respiratory distress assessment (nasal flaring, retractions, grunting).
2. Laboratory & Imaging Tests (For Severe Cases Only)
- Nasopharyngeal Swab (PCR or Immunofluorescence):
- Detects RSV or other viruses.
- Pulse Oximetry:
- Measures oxygen saturation.
- Chest X-ray (Not Routine, Used if Severe Case):
- Hyperinflation, peribronchial thickening, patchy atelectasis.
- Blood Gas Analysis (ABG):
- Assesses hypoxia & CO₂ retention.
- CBC (If Bacterial Superinfection Suspected):
- Mild leukocytosis in secondary bacterial infection.
Medical Management:
There is no specific antiviral treatment for bronchiolitis. Supportive care is the mainstay of treatment.
1. Home Management (For Mild Cases)
- Nasal Suctioning with Saline Drops: Clears nasal congestion.
- Hydration & Nutrition: Small frequent feeds to prevent dehydration.
- Antipyretics (Paracetamol/Ibuprofen): Controls fever.
- Humidified Air: Reduces airway irritation.
2. Hospital Management (For Moderate to Severe Cases)
A. Respiratory Support
- Oxygen Therapy:
- If SpO₂ < 92% or signs of severe distress.
- Nasal cannula or face mask for mild cases.
- High-flow nasal cannula (HFNC) for moderate cases.
- Non-invasive ventilation (CPAP/BiPAP) if respiratory failure risk.
- Nebulization (Only for Severe Bronchospasm)
- Hypertonic Saline Nebulization (3%): Helps loosen mucus.
- Bronchodilators (Salbutamol, Epinephrine nebulization):
- Only beneficial if the child has underlying asthma or wheezing.
- Corticosteroids (Dexamethasone/Prednisolone)
- Not routinely recommended unless severe inflammation.
- Intravenous Fluids (IVF)
- If poor feeding or dehydration is present.
- Antiviral Therapy
- Ribavirin (Antiviral for RSV) is rarely used (only in immunocompromised infants).
- Antibiotics (ONLY if bacterial pneumonia is suspected)
- Amoxicillin-clavulanate or Azithromycin.
Surgical Management:
- Surgery is NOT required for bronchiolitis.
- Tracheostomy or mechanical ventilation is needed only in severe respiratory failure.
Nursing Management:
1. Assessment:
- Monitor respiratory rate & work of breathing (retractions, nasal flaring).
- Check oxygen saturation (SpO₂).
- Assess hydration status (urine output, mucous membrane dryness).
- Monitor for cyanosis & lethargy (signs of worsening hypoxia).
2. Nursing Interventions:
A. Airway & Breathing Support
- Positioning: Infant in semi-upright (30-45°) position to reduce airway obstruction.
- Nasal Suctioning: Frequent clearing of mucus for easier breathing.
- Administer Oxygen Therapy if SpO₂ <92%.
- Nebulization Therapy (if prescribed):
- Hypertonic saline nebulization helps loosen mucus.
- Bronchodilators (Salbutamol/Epinephrine) only in select cases.
B. Hydration & Nutrition
- Encourage breastfeeding or small frequent feeds.
- IV Fluids if oral feeding is not possible.
C. Fever & Comfort Management
- Administer antipyretics (Paracetamol, Ibuprofen).
- Provide humidified air or steam inhalation.
D. Parental Education & Discharge Instructions
- Educate caregivers on symptom monitoring:
- Signs of worsening distress (cyanosis, difficulty feeding).
- When to seek emergency care.
- Hand Hygiene & Infection Control:
- RSV is highly contagious, spread through direct contact & droplets.
- Avoid passive smoke exposure.
- Vaccination:
- Palivizumab (Synagis) for high-risk infants (preterm, heart disease).
Complications of Bronchiolitis:
- Respiratory Failure (if severe airway obstruction).
- Apnea (Common in preterm infants).
- Dehydration due to poor feeding.
- Secondary bacterial pneumonia.
- Recurrent wheezing/asthma in some children.
Prognosis:
- Most cases resolve within 7-10 days.
- Hospitalization required in severe cases.
- Recurrent wheezing may persist in some children.
Key Points:
✔ Bronchiolitis is a viral infection causing inflammation of small airways in infants.
✔ RSV is the most common cause.
✔ Oxygen therapy & hydration are the main treatments.
✔ Antibiotics are not required unless a bacterial infection is present.
✔ Prevention includes hand hygiene & Palivizumab for high-risk infants.
Pneumonia in Children
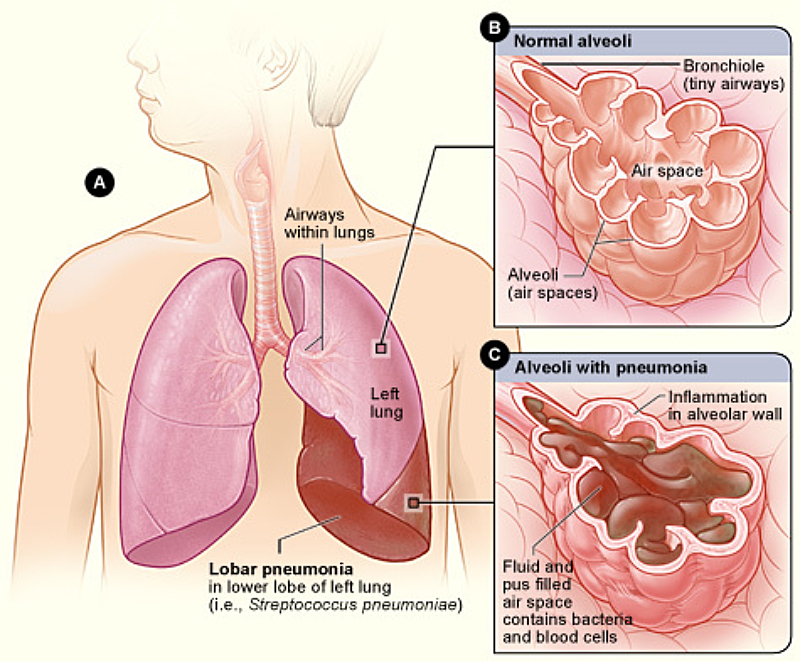
Definition:
Pneumonia is an acute inflammatory infection of the lungs, affecting the alveoli (air sacs) and resulting in fluid or pus accumulation. It leads to difficulty breathing, fever, cough, and chest pain. Pneumonia can be caused by bacteria, viruses, fungi, or aspiration of foreign substances.
Etiology (Causes):
Pneumonia can be classified based on the causative agent.
1. Viral Pneumonia (Most Common in Children – 60-80%)
- Respiratory Syncytial Virus (RSV)
- Influenza virus (A & B)
- Parainfluenza virus
- Adenovirus
- Human Metapneumovirus
- Coronavirus (Including COVID-19)
2. Bacterial Pneumonia (More Severe, Less Common in Children)
- Streptococcus pneumoniae (Most common bacterial cause)
- Haemophilus influenzae type B (Hib)
- Staphylococcus aureus
- Mycoplasma pneumoniae (Atypical pneumonia, common in older children)
- Klebsiella pneumoniae
- Pseudomonas aeruginosa
3. Fungal Pneumonia (Rare)
- Pneumocystis jirovecii (in immunocompromised children, e.g., HIV)
- Histoplasma, Aspergillus
4. Aspiration Pneumonia
- Occurs when foreign substances (food, liquids, vomit) enter the lungs.
- Common in children with swallowing disorders, gastroesophageal reflux disease (GERD), or neurologic conditions.
Risk Factors:
- Age: Children under 5 years, especially infants.
- Malnutrition & vitamin A deficiency.
- Preterm birth & low birth weight.
- Weakened immune system (e.g., HIV, immunosuppressive therapy).
- Chronic illnesses (asthma, cystic fibrosis, congenital heart disease).
- Exposure to tobacco smoke, air pollution.
- Lack of breastfeeding (passive immunity loss).
Types of Pneumonia:
1. Community-Acquired Pneumonia (CAP)
- Develops outside the hospital.
- Caused by viral or bacterial pathogens.
2. Hospital-Acquired Pneumonia (HAP)
- Occurs in hospitalized children (≥48 hours after admission).
- Often more severe, antibiotic-resistant pathogens.
3. Aspiration Pneumonia
- Due to inhalation of foreign substances.
- Common in children with swallowing difficulties or reflux.
4. Atypical Pneumonia (Walking Pneumonia)
- Mild symptoms, often caused by Mycoplasma pneumoniae.
- More common in older children & adolescents.
Pathophysiology:
- Pathogen Entry: Infection enters the lungs via inhalation, aspiration, or bloodstream.
- Inflammatory Response:
- Alveolar inflammation leads to fluid & pus accumulation.
- Mucosal edema & mucus production cause airway obstruction.
- Impaired Gas Exchange:
- Oxygenation decreases due to fluid-filled alveoli.
- Leads to hypoxia (low oxygen levels) & respiratory distress.
- Severe Cases (Complications):
- Pleural effusion (fluid in pleural space)
- Lung abscess
- Septicemia (infection spreading in blood).
Clinical Manifestations (Signs & Symptoms):
Mild to Moderate Pneumonia:
- Fever (38-40°C or 100.4-104°F)
- Cough (dry or productive)
- Fast breathing (Tachypnea):
- >50 breaths/min (Infants 2-12 months)
- >40 breaths/min (Children 1-5 years)
- Nasal flaring & intercostal retractions (difficulty breathing)
- Crackles or decreased breath sounds on auscultation
- Lethargy, poor feeding, irritability
- Sweating, chills
Severe Pneumonia (Emergency Symptoms):
- Cyanosis (blue lips, fingers, tongue)
- Severe difficulty breathing (grunting, nasal flaring, head bobbing)
- Chest indrawing (ribs pulling in with breathing)
- Altered mental status (lethargy, seizures)
- Persistent vomiting, dehydration signs
- Shock (low blood pressure, cold extremities)
Diagnosis:
1. Clinical Examination:
- Auscultation: Crackles, rhonchi, decreased breath sounds.
- Chest Percussion: Dullness over lung areas (fluid accumulation).
2. Laboratory & Imaging Tests:
- Chest X-ray: Confirms pneumonia type (lobar, interstitial, pleural effusion).
- Complete Blood Count (CBC):
- High WBCs (bacterial pneumonia)
- Lymphocytosis (viral pneumonia)
- Blood Cultures: Identifies bacteria in severe pneumonia.
- Sputum Culture: Identifies bacterial infection.
- Pulse Oximetry: Measures oxygen saturation (SpO₂ <92% = Severe pneumonia).
- Arterial Blood Gas (ABG): Assesses hypoxia, respiratory acidosis.
Medical Management:
1. Supportive Therapy (For Viral & Mild Bacterial Pneumonia)
- Oxygen Therapy: If SpO₂ <92%.
- Antipyretics (Paracetamol, Ibuprofen): For fever & discomfort.
- Hydration: IV fluids if oral intake is poor.
- Nasal suctioning: Clears secretions in infants.
- Nutritional Support: Frequent, small feeds.
2. Antibiotic Therapy (For Bacterial Pneumonia)
- First-line (Oral, Mild Cases):
- Amoxicillin (High Dose).
- Severe Cases (IV Antibiotics):
- Ceftriaxone or Cefotaxime + Azithromycin.
- Atypical Pneumonia:
- Macrolides (Azithromycin, Clarithromycin).
3. Antiviral Therapy (For Influenza Pneumonia)
- Oseltamivir (Tamiflu) for Influenza A/B within 48 hours of symptoms.
4. Bronchodilators (For Wheezing or Bronchospasm)
- Salbutamol (Albuterol) Nebulization.
Surgical Management:
Surgery is not required unless complications arise:
- Chest Tube Drainage (If pleural effusion/empyema).
- Thoracotomy (Lung abscess drainage, rare cases).
- Mechanical Ventilation (Severe respiratory failure).
Nursing Management:
1. Assessment:
- Monitor respiratory rate & effort (nasal flaring, retractions, grunting).
- Assess oxygen saturation (SpO₂) & signs of cyanosis.
- Monitor fever, hydration status (urine output, mucous membranes).
- Assess auscultation findings (wheezing, crackles, diminished breath sounds).
2. Nursing Interventions:
A. Airway & Breathing Support
- Administer oxygen therapy if SpO₂ <92%.
- Positioning: Semi-Fowler’s or upright to ease breathing.
- Suction secretions in infants (gentle nasal suctioning).
- Administer prescribed bronchodilators (if wheezing present).
B. Fever & Hydration Management
- Administer antipyretics (Paracetamol/Ibuprofen).
- Encourage oral fluids or IV hydration.
- Monitor for dehydration (dry lips, poor skin turgor).
C. Nutrition Support
- Encourage small, frequent meals.
- NG tube feeding (if severe difficulty in feeding).
D. Infection Control & Education
- Encourage hand hygiene & vaccination (PCV, Hib, Influenza).
- Avoid smoking exposure.
- Teach parents about warning signs (cyanosis, chest indrawing, high fever).
Complications:
- Pleural Effusion/Empyema
- Lung Abscess
- Sepsis (Bacterial Spread)
- Respiratory Failure
Prognosis:
- Mild pneumonia recovers in 7-10 days.
- Severe cases may require hospitalization but have a good outcome if treated promptly.
Cardiovascular system:
Atrial Septal Defect (ASD)
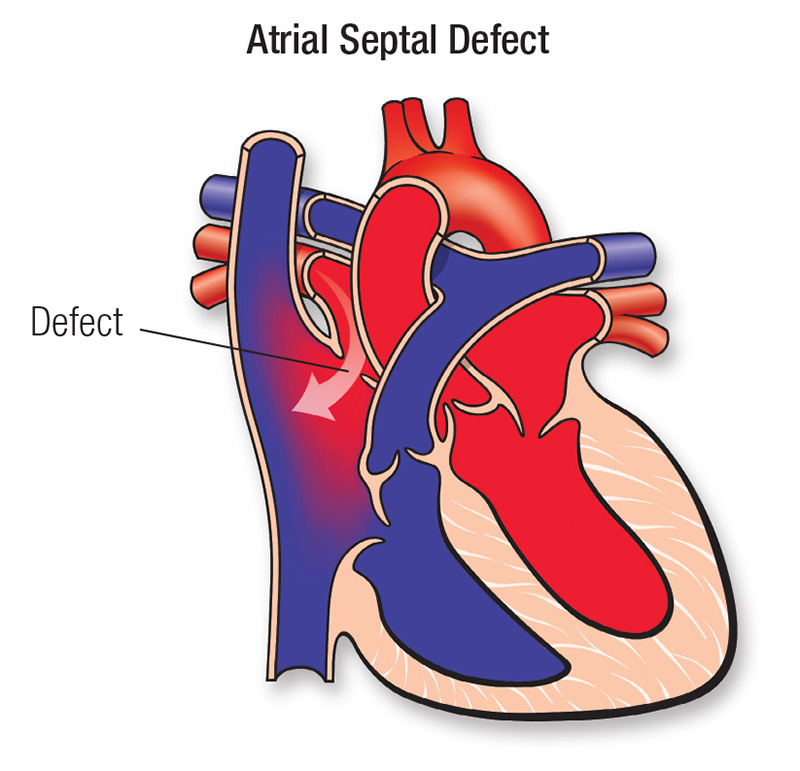
Definition:
Atrial Septal Defect (ASD) is a congenital heart defect characterized by an abnormal opening in the atrial septum, allowing oxygenated blood from the left atrium to mix with deoxygenated blood in the right atrium. This leads to increased pulmonary blood flow, volume overload, and right-sided heart strain.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Familial inheritance of congenital heart defects.
- Chromosomal abnormalities:
- Down Syndrome (Trisomy 21)
- Holt-Oram Syndrome (ASD + limb abnormalities)
- Noonan Syndrome
- Ellis-van Creveld Syndrome
2. Environmental Factors:
- Maternal infections (Rubella during pregnancy)
- Maternal diabetes
- Maternal smoking, alcohol, or drug use
- Advanced maternal age (>35 years)
Types of ASD:
- Ostium Secundum (Most Common – 70%)
- Located in the middle of the atrial septum.
- Can close spontaneously in small defects.
- Ostium Primum (20%)
- Located near the lower part of the atrial septum.
- Often associated with endocardial cushion defects & Down Syndrome.
- Sinus Venosus ASD (10%)
- Located near the superior vena cava (SVC) or inferior vena cava (IVC).
- Associated with abnormal pulmonary venous drainage.
- Coronary Sinus ASD (Rare)
- Defect near the coronary sinus.
- Often accompanied by persistent left superior vena cava.
Pathophysiology:
- Abnormal opening in the atrial septum allows left-to-right shunting of blood.
- Increased blood volume in the right atrium → right ventricular volume overload.
- Increased pulmonary blood flow leads to:
- Pulmonary hypertension (in severe, untreated cases).
- Right atrial & ventricular dilation.
- Over time, right-sided heart failure may develop due to chronic volume overload.
- If untreated for many years → Eisenmenger syndrome (reversal of shunt, cyanosis, right-to-left shunting).
Clinical Manifestations (Signs & Symptoms):
Small ASD (≤5mm)
- Asymptomatic (may close spontaneously).
- Incidental murmur detected on routine check-up.
Moderate to Large ASD (>5mm)
- Frequent respiratory infections (recurrent pneumonia, bronchitis).
- Failure to thrive (poor weight gain)
- Exercise intolerance (easy fatigue, shortness of breath)
- Mild cyanosis (if right-to-left shunt develops)
- Palpitations or arrhythmias (Atrial fibrillation, Atrial flutter).
- Right-sided heart failure (if untreated) → Peripheral edema, hepatomegaly.
- Heart murmur (systolic ejection murmur at the pulmonary area).
Severe or Late ASD (With Eisenmenger Syndrome)
- Cyanosis & clubbing (right-to-left shunting)
- Loud P2 (Pulmonary hypertension)
- Arrhythmias due to atrial dilation
- Stroke (paradoxical embolism due to right-to-left shunting)
Diagnosis:
1. Physical Examination:
- Wide, fixed split of the second heart sound (S2)
- Systolic ejection murmur at the left upper sternal border
- Diastolic murmur (if large defect causes increased pulmonary flow)
2. Imaging & Laboratory Tests:
- Echocardiography (Gold Standard)
- Shows atrial septal defect & shunting of blood.
- Bubble contrast study detects right-to-left shunt.
- Chest X-ray
- Cardiomegaly (enlarged right atrium/ventricle).
- Increased pulmonary vascular markings.
- Electrocardiogram (ECG)
- Right axis deviation (RAD) due to right ventricular overload.
- Right bundle branch block (RBBB).
- Cardiac MRI/CT Scan
- Used in complex cases to assess pulmonary veins & defect location.
- Cardiac Catheterization
- Measures oxygen saturation & pressure differences between right & left atrium.
- Confirms pulmonary hypertension (if present).
Medical Management:
1. Conservative Management (For Small Defects)
- Observation & follow-up echocardiograms (small ASDs <5mm may close spontaneously).
- Endocarditis prophylaxis is NOT needed for ASD alone.
- Preventive antibiotics if associated with a prosthetic heart valve or surgical patch.
2. Medications (For Symptom Management)
- Diuretics (Furosemide) – Reduces fluid overload in heart failure cases.
- ACE Inhibitors (Enalapril, Captopril) – Helps reduce pulmonary hypertension.
- Beta-blockers (Propranolol, Atenolol) – Controls atrial arrhythmias.
- Anticoagulants (Aspirin, Warfarin) – For patients with atrial fibrillation to prevent stroke.
Surgical Management:
1. Indications for Surgery
- ASD >5mm with significant left-to-right shunting.
- Symptoms of heart failure (exercise intolerance, recurrent infections).
- Pulmonary hypertension developing due to shunting.
- Right atrial/ventricular enlargement seen on echocardiography.
2. Types of Surgical Interventions
A. Catheter-Based Closure (Preferred for Ostium Secundum ASD)
- Transcatheter device closure (Amplatzer Septal Occluder):
- Minimally invasive (via femoral vein).
- Suitable for ASD <35mm with adequate rim.
- Not suitable for Ostium Primum or Sinus Venosus ASD.
B. Open-Heart Surgery (For Large or Complex ASDs)
- Direct suture closure (for small defects).
- Pericardial or synthetic patch closure (for large ASDs).
- Performed under cardiopulmonary bypass.
Nursing Management:
1. Preoperative Nursing Care:
- Assess respiratory function: Monitor for tachypnea, retractions.
- Monitor for signs of heart failure: Peripheral edema, hepatomegaly.
- Administer prescribed medications (diuretics, beta-blockers).
- Maintain fluid balance: Prevent volume overload.
- Educate parents about surgery & recovery expectations.
2. Postoperative Nursing Care:
- Monitor vital signs & cardiac status:
- Watch for arrhythmias, hypotension, bleeding.
- Chest tube care (if open-heart surgery performed).
- Pain management (IV analgesics in the first 24 hours).
- Encourage early mobilization to prevent blood clots.
- Monitor for signs of infection at surgical site.
- Antibiotic prophylaxis (if a prosthetic patch is used).
- Gradual reintroduction to normal feeding & activity.
- Provide emotional support & discharge teaching.
Complications of ASD:
- Paradoxical Embolism → Stroke (If right-to-left shunting develops).
- Eisenmenger Syndrome (Irreversible pulmonary hypertension).
- Atrial Arrhythmias (Atrial fibrillation, Atrial flutter).
- Congestive Heart Failure (Right-sided failure in late cases).
- Pulmonary Hypertension (Due to chronic left-to-right shunting).
Prognosis:
- Small ASDs may close spontaneously by age 2-5 years.
- Surgical closure has a >95% success rate with excellent prognosis.
- Late diagnosis & untreated cases can lead to pulmonary hypertension & heart failure.
Key Points:
✔ ASD causes left-to-right shunting, increasing pulmonary blood flow.
✔ Small ASDs may close spontaneously; large ASDs require closure.
✔ Surgical or catheter-based closure is effective with a high success rate.
✔ Nursing care focuses on monitoring heart function, preventing complications, and family education.
Ventricular Septal Defect (VSD)
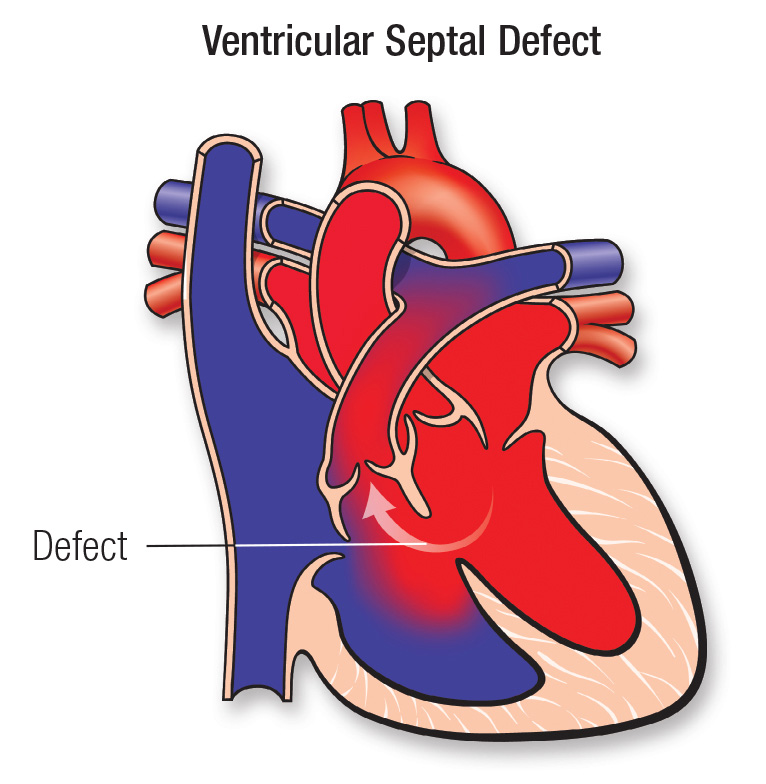
Definition:
Ventricular Septal Defect (VSD) is a congenital heart defect characterized by an abnormal opening in the ventricular septum, allowing oxygenated blood from the left ventricle (LV) to mix with deoxygenated blood in the right ventricle (RV). This left-to-right shunting increases pulmonary blood flow, leading to pulmonary hypertension, heart failure, and right ventricular hypertrophy if untreated.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Familial inheritance of congenital heart diseases.
- Chromosomal abnormalities:
- Down Syndrome (Trisomy 21).
- DiGeorge Syndrome (22q11 deletion).
- Turner Syndrome.
- Holt-Oram Syndrome (Heart-hand syndrome).
2. Environmental Factors:
- Maternal infections (Rubella, Cytomegalovirus, Toxoplasmosis).
- Maternal diabetes (Gestational & Pre-existing Type 1 or 2).
- Maternal alcohol, drug use (e.g., anticonvulsants, warfarin, lithium).
- Exposure to radiation, teratogens during pregnancy.
Types of VSD:
- Perimembranous VSD (Most Common – 80%)
- Located in the upper part of the septum (near aortic valve).
- May close spontaneously in early childhood.
- Muscular VSD (15-20%)
- Located in the lower muscular septum.
- Small defects may close spontaneously.
- Inlet VSD (5-10%)
- Located near the tricuspid and mitral valves.
- Associated with atrioventricular septal defects (AVSDs), Down Syndrome.
- Supracristal (Outlet) VSD (Rare – <5%)
- Located beneath the pulmonary valve.
- High risk of aortic valve prolapse and regurgitation.
Pathophysiology:
- Abnormal opening in the ventricular septum allows oxygenated blood from the left ventricle (high pressure) to mix with deoxygenated blood in the right ventricle (low pressure).
- Increased pulmonary blood flow due to left-to-right shunting.
- Volume overload in the lungs → Leads to pulmonary congestion & edema.
- Right ventricular hypertrophy (RVH) due to increased workload.
- Increased pulmonary vascular resistance (PVR) → If untreated, Eisenmenger Syndrome (Right-to-left shunt, cyanosis, pulmonary hypertension) develops.
- Heart failure (CHF) in large VSDs due to excessive volume overload.
Clinical Manifestations (Signs & Symptoms):
Small VSD (<5mm)
- Asymptomatic (may close spontaneously by 1-2 years).
- Murmur detected incidentally during routine check-up.
- Harsh holosystolic murmur at the left lower sternal border.
Moderate to Large VSD (>5mm)
- Frequent respiratory infections (pneumonia, bronchitis).
- Failure to thrive (poor weight gain, difficulty feeding).
- Tachypnea, tachycardia, dyspnea (increased work of breathing).
- Sweating while feeding (due to increased cardiac workload).
- Palpable thrill (vibratory sensation on the chest wall).
- Hepatomegaly (liver enlargement in heart failure cases).
- Loud, harsh pansystolic murmur at the left lower sternal border.
Severe VSD (With Eisenmenger Syndrome)
- Cyanosis (bluish lips, skin, clubbing of fingers/toes).
- Polycythemia (increased red blood cell count to compensate for hypoxia).
- Dyspnea on exertion.
- Arrhythmias & syncope.
- Right heart failure (edema, hepatomegaly, jugular venous distension – JVD).
Diagnosis:
1. Physical Examination:
- Loud, harsh holosystolic murmur at the left lower sternal border.
- Palpable thrill (vibratory murmur).
- Signs of heart failure (tachycardia, hepatomegaly, poor feeding).
2. Imaging & Laboratory Tests:
- Echocardiography (Gold Standard)
- Detects size, location of VSD.
- Identifies left-to-right shunting.
- Measures pulmonary artery pressure.
- Chest X-ray
- Cardiomegaly (Enlarged left & right ventricles).
- Increased pulmonary vascular markings (pulmonary congestion).
- Electrocardiogram (ECG)
- Left ventricular hypertrophy (LVH) & Right ventricular hypertrophy (RVH).
- Right axis deviation in severe VSD.
- Cardiac MRI/CT Scan
- Used in complex cases for detailed visualization.
- Cardiac Catheterization
- Measures pulmonary artery pressure.
- Identifies pulmonary hypertension (Eisenmenger Syndrome).
Medical Management:
1. Conservative Management (For Small VSDs)
- Observation & periodic echocardiograms.
- Most small muscular VSDs close spontaneously by 2-5 years.
- Prophylactic antibiotics NOT needed unless high-risk for endocarditis.
2. Medications (For Symptomatic or Moderate VSDs)
- Diuretics (Furosemide, Spironolactone) – Reduces pulmonary congestion & heart failure symptoms.
- ACE Inhibitors (Enalapril, Captopril) – Lowers pulmonary hypertension.
- Digoxin – Improves cardiac contractility in heart failure cases.
- High-calorie nutrition support – Helps weight gain in failure to thrive infants.
Surgical Management:
1. Indications for Surgery
- Large VSD (>5-10mm) with heart failure symptoms.
- Pulmonary hypertension due to excessive left-to-right shunting.
- Failure to thrive despite medical therapy.
- Right ventricular dilation or dysfunction.
- Recurrent pneumonia or frequent infections.
2. Types of Surgical Interventions
A. Catheter-Based Device Closure (For Muscular VSD)
- Transcatheter closure (Amplatzer Muscular VSD Occluder).
- Minimally invasive (via femoral vein).
- NOT suitable for perimembranous or inlet VSDs.
B. Open-Heart Surgery (Preferred for Large VSDs)
- Direct suture closure (For small defects).
- Pericardial or synthetic patch closure (For large defects).
- Performed under cardiopulmonary bypass.
Nursing Management:
1. Preoperative Nursing Care:
- Monitor respiratory function: Tachypnea, retractions, cyanosis.
- Assess for signs of heart failure: Hepatomegaly, poor feeding.
- Administer prescribed medications (diuretics, digoxin, ACE inhibitors).
- Maintain fluid balance: Prevent volume overload.
- Educate parents about surgical procedure & recovery.
2. Postoperative Nursing Care:
- Monitor vital signs & cardiac status:
- Assess for arrhythmias, hypotension, bleeding.
- Chest tube care (if open-heart surgery performed).
- Pain management (IV analgesics in the first 24 hours).
- Encourage early mobilization to prevent blood clots.
- Monitor for infection at surgical site.
- Antibiotic prophylaxis (if a prosthetic patch is used).
- Gradual reintroduction to normal feeding & activity.
- Provide emotional support & discharge teaching.
Complications of VSD:
- Pulmonary Hypertension & Eisenmenger Syndrome.
- Congestive Heart Failure (CHF).
- Aortic Valve Prolapse & Regurgitation.
- Arrhythmias (Atrial fibrillation, Ventricular tachycardia).
- Infective Endocarditis.
Prognosis:
- Small VSDs may close spontaneously by 5 years.
- Surgical closure has a >95% success rate.
- Early closure prevents long-term complications.
Key Points:
✔ VSD causes left-to-right shunting, increasing pulmonary blood flow.
✔ Small VSDs may close spontaneously; large VSDs require surgical repair.
✔ Surgical closure is highly successful, preventing complications.
✔ Nursing care focuses on monitoring heart function, preventing complications, and family education.
Patent Ductus Arteriosus (PDA)
Definition:
Patent Ductus Arteriosus (PDA) is a congenital heart defect where the ductus arteriosus, a fetal blood vessel connecting the pulmonary artery to the aorta, fails to close after birth. This results in abnormal left-to-right shunting of blood, causing increased pulmonary blood flow, volume overload, and heart failure if left untreated.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Family history of congenital heart disease.
- Chromosomal disorders:
- Down Syndrome (Trisomy 21).
- Rubella Syndrome.
- Char Syndrome (associated with PDA and facial anomalies).
2. Environmental Factors:
- Prematurity (Most Common Cause).
- Maternal Rubella Infection during first trimester.
- High-altitude births (Low oxygen levels delay closure).
- Neonatal hypoxia or respiratory distress syndrome (RDS).
- Exposure to teratogens (alcohol, phenytoin, NSAIDs during pregnancy).
Types of PDA:
- Small PDA (<1.5mm)
- Minimal shunting, often asymptomatic.
- May close spontaneously within the first year.
- Moderate PDA (1.5-4mm)
- Leads to murmur, mild heart failure symptoms.
- May require medical or catheter-based closure.
- Large PDA (>4mm)
- Significant left-to-right shunting.
- Causes pulmonary hypertension & heart failure.
- Needs early surgical intervention.
Pathophysiology:
- In fetal life, the ductus arteriosus remains open to allow blood to bypass the non-functioning lungs.
- After birth, due to oxygen increase & prostaglandin E2 decrease, the ductus arteriosus closes within 24-48 hours.
- If PDA remains open:
- Left-to-right shunting occurs (Aorta → Pulmonary artery).
- Increased pulmonary blood flow → Leads to pulmonary congestion & edema.
- Left atrium & ventricle volume overload → Can cause left ventricular hypertrophy (LVH).
- Progressive pulmonary hypertension.
- If untreated, Eisenmenger Syndrome develops (right-to-left shunting, cyanosis, clubbing).
Clinical Manifestations (Signs & Symptoms):
Small PDA (Asymptomatic)
- Incidental murmur on routine check-up.
- May close spontaneously within the first year.
Moderate to Large PDA
- Continuous “Machinery” Murmur (Left upper sternal border).
- Bounding peripheral pulses.
- Wide pulse pressure (Systolic BP ↑, Diastolic BP ↓).
- Frequent respiratory infections (Pneumonia, Bronchitis).
- Failure to thrive (Poor weight gain, Feeding difficulties).
- Tachypnea, tachycardia, sweating while feeding.
- Hepatomegaly (If heart failure develops).
Severe PDA (With Eisenmenger Syndrome)
- Cyanosis & Clubbing of fingers/toes.
- Polycythemia (Increased RBC count due to chronic hypoxia).
- Dyspnea on exertion.
- Right heart failure symptoms (Edema, Hepatomegaly, JVD).
Diagnosis:
1. Physical Examination:
- Continuous “Machine-like” Murmur at left upper sternal border.
- Bounding peripheral pulses.
- Wide pulse pressure.
2. Imaging & Laboratory Tests:
- Echocardiography (Gold Standard)
- Identifies abnormal shunting (left-to-right flow).
- Measures PDA size & pulmonary artery pressure.
- Chest X-ray
- Cardiomegaly (Enlarged left atrium & left ventricle).
- Increased pulmonary vascular markings.
- Electrocardiogram (ECG)
- Left ventricular hypertrophy (LVH).
- Right ventricular hypertrophy (RVH) in Eisenmenger syndrome.
- Cardiac MRI/CT Scan
- Used for complex cases to assess PDA anatomy.
- Cardiac Catheterization
- Measures oxygen saturation & pulmonary hypertension.
Medical Management:
1. Conservative Management (For Small PDAs)
- Observation & periodic echocardiograms (Many close spontaneously in preterm infants).
- Endocarditis prophylaxis NOT needed unless high-risk for infection.
2. Medications (For Premature Infants)
- Prostaglandin Inhibitors (NSAIDs)
- Indomethacin or Ibuprofen:
- Stimulates ductal closure in preterm infants.
- Not effective for full-term infants or large PDAs.
- Indomethacin or Ibuprofen:
- Diuretics (Furosemide)
- Reduces pulmonary congestion & heart failure symptoms.
- ACE Inhibitors (Captopril, Enalapril)
- Decreases left ventricular overload & lowers BP.
- High-Calorie Nutrition Support
- Helps weight gain in failure-to-thrive infants.
Surgical Management:
1. Indications for Surgery
- Large PDA (>4mm) with significant shunting.
- Pulmonary hypertension or heart failure symptoms.
- Failure of medical therapy (Indomethacin non-responsive PDA).
- Persistent PDA beyond infancy.
2. Types of Surgical Interventions
A. Catheter-Based Device Closure (Preferred for Small to Moderate PDA)
- Transcatheter occlusion using PDA closure devices (Amplatzer, Coil embolization).
- Minimally invasive (via femoral artery/vein).
- Short hospital stay, fast recovery.
B. Surgical Ligation (For Large PDA or Premature Infants)
- Open thoracotomy or Video-Assisted Thoracoscopic Surgery (VATS).
- Ductus arteriosus is ligated or clipped to prevent shunting.
- Indicated if PDA is too large for catheter-based closure.
Nursing Management:
1. Preoperative Nursing Care:
- Monitor respiratory function: Tachypnea, retractions, cyanosis.
- Assess for signs of heart failure: Hepatomegaly, poor feeding.
- Administer prescribed medications (Diuretics, NSAIDs, ACE inhibitors).
- Maintain fluid balance: Prevent volume overload.
- Educate parents about the procedure & post-op care.
2. Postoperative Nursing Care:
- Monitor vital signs & cardiac status:
- Check for arrhythmias, hypotension, bleeding.
- Monitor for signs of pulmonary complications (Atelectasis, pneumonia).
- Pain management (IV analgesics for first 24 hours).
- Encourage early mobilization to prevent lung complications.
- Monitor surgical site for infection.
- Antibiotic prophylaxis (if prosthetic closure device used).
- Gradual return to normal feeding & activity.
- Provide emotional support & discharge education.
Complications of PDA:
- Heart Failure (CHF) due to volume overload.
- Pulmonary Hypertension & Eisenmenger Syndrome.
- Endocarditis (Infection of heart lining).
- Arrhythmias (Atrial fibrillation, Ventricular tachycardia).
- Failure to thrive (Poor growth in infants).
Prognosis:
- Small PDAs may close spontaneously by 1-2 years.
- Medical & surgical closure has a >98% success rate.
- Early intervention prevents long-term complications.
Key Points:
✔ PDA causes left-to-right shunting, increasing pulmonary blood flow.
✔ Premature infants often respond to NSAIDs (Indomethacin/Ibuprofen).
✔ Large PDAs require catheter-based closure or surgical ligation.
✔ Early closure prevents heart failure & pulmonary hypertension.
✔ Nursing care focuses on monitoring cardiac function, preventing complications, and family education.
Tetralogy of Fallot (TOF)
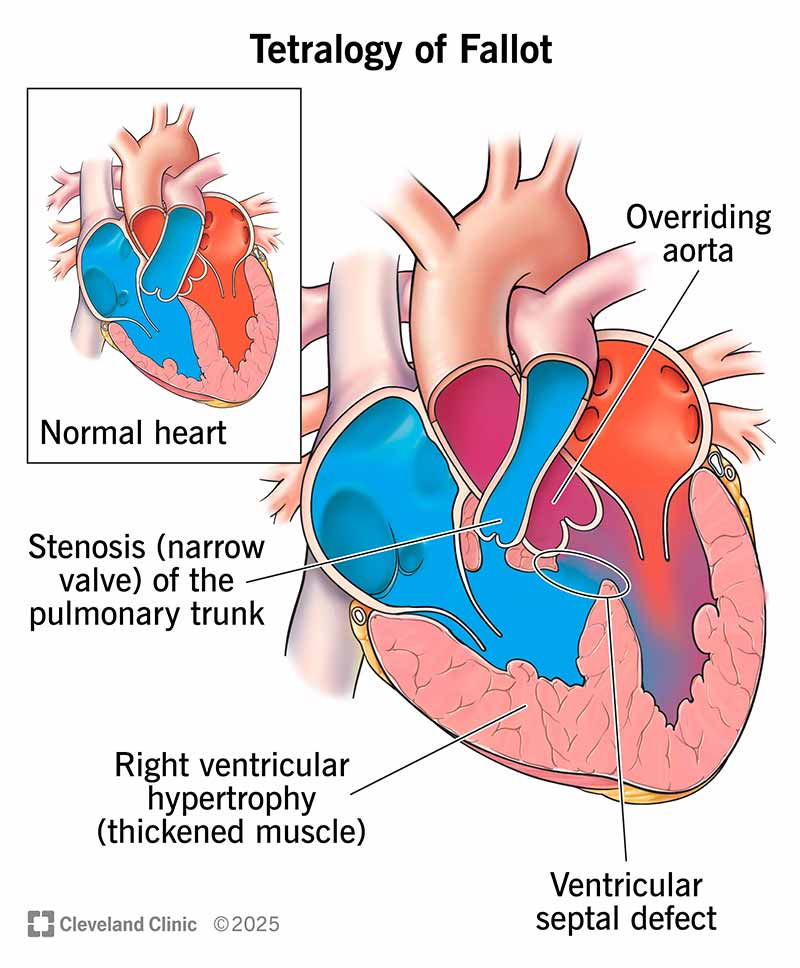
Definition:
Tetralogy of Fallot (TOF) is a cyanotic congenital heart defect characterized by a combination of four heart anomalies that result in poor oxygenation of blood, leading to cyanosis (bluish skin discoloration), hypoxia, and right ventricular hypertrophy.
The four components of TOF are:
- Pulmonary Stenosis – Narrowing of the pulmonary valve or artery reduces blood flow to the lungs.
- Ventricular Septal Defect (VSD) – A hole between the right and left ventricles allows mixing of oxygenated and deoxygenated blood.
- Overriding Aorta – The aorta is positioned over both the left and right ventricles, receiving mixed blood.
- Right Ventricular Hypertrophy (RVH) – The right ventricle thickens due to increased workload.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Chromosomal abnormalities:
- 22q11 deletion syndrome (DiGeorge Syndrome)
- Down Syndrome (Trisomy 21)
- Alagille Syndrome
- Noonan Syndrome
- Family history of congenital heart disease.
2. Environmental Factors:
- Maternal Rubella infection during pregnancy.
- Maternal diabetes (poorly controlled).
- Exposure to teratogens (alcohol, drugs, retinoic acid, thalidomide).
- Advanced maternal age (>40 years).
- Poor maternal nutrition (folic acid deficiency).
Types of TOF:
1. Classic Tetralogy of Fallot
- Most common type with all four defects.
2. TOF with Pulmonary Atresia
- Complete obstruction of the pulmonary valve, requiring a Patent Ductus Arteriosus (PDA) or collateral vessels to supply blood to the lungs.
3. TOF with Absent Pulmonary Valve
- Pulmonary valve is underdeveloped or absent, causing severe pulmonary regurgitation and airway compression.
4. TOF with Double Outlet Right Ventricle (DORV)
- Aorta and pulmonary artery both originate from the right ventricle, worsening cyanosis.
Pathophysiology:
- Pulmonary Stenosis reduces blood flow to the lungs, leading to decreased oxygenation.
- Right Ventricular Hypertrophy occurs due to increased workload against the stenosed pulmonary artery.
- Ventricular Septal Defect (VSD) allows mixing of oxygenated and deoxygenated blood, leading to systemic hypoxia.
- Overriding Aorta receives mixed blood, resulting in cyanosis (low oxygen levels in blood).
- “Tet Spells” (Hypercyanotic Episodes) occur due to increased right-to-left shunting, causing severe hypoxia and syncope.
Clinical Manifestations (Signs & Symptoms):
Mild TOF (Less Severe Pulmonary Stenosis)
- Mild cyanosis at birth or delayed onset (few months).
- Murmur detected during routine check-up.
- Poor weight gain, fatigue, delayed milestones.
Severe TOF (Severe Pulmonary Stenosis)
- Cyanosis (bluish skin, lips, fingers, toes).
- Dyspnea on exertion (difficulty breathing while feeding, crying, or playing).
- “Tet Spells” (Hypercyanotic Episodes):
- Sudden worsening of cyanosis.
- Child squats instinctively to increase blood flow to the lungs.
- Severe breathlessness, unconsciousness, seizures in extreme cases.
- Clubbing of fingers and toes (due to chronic hypoxia).
- Loud systolic murmur at the left upper sternal border.
- Polycythemia (Increased RBC production due to chronic hypoxia).
- Growth retardation, developmental delay.
Diagnosis:
1. Physical Examination:
- Cyanosis (bluish skin, lips, and extremities).
- Clubbing of fingers & toes.
- Harsh systolic ejection murmur (Left upper sternal border).
- Tachypnea & “Tet Spells”.
2. Imaging & Laboratory Tests:
- Echocardiography (Gold Standard)
- Identifies pulmonary stenosis, VSD, overriding aorta, and RVH.
- Measures pressure gradients across the pulmonary valve.
- Chest X-ray
- “Boot-shaped heart” due to right ventricular hypertrophy.
- Decreased pulmonary vascular markings (reduced blood flow to lungs).
- Electrocardiogram (ECG)
- Right Axis Deviation.
- Right Ventricular Hypertrophy (RVH).
- Cardiac MRI/CT Scan
- Used in complex cases for surgical planning.
- Pulse Oximetry
- Low oxygen saturation (<85%) indicates cyanosis.
- Blood Tests
- Polycythemia (Increased Hematocrit & RBC count).
- Cardiac Catheterization
- Measures oxygen levels & pressure gradients in the heart.
Medical Management:
1. Conservative Management (Pre-Surgery)
- Prostaglandin E1 Infusion (If PDA is needed for pulmonary blood flow).
- Oxygen Therapy (Limited effect as cyanosis is due to shunting).
- IV Fluids & Electrolyte Management (Prevent dehydration & polycythemia).
- Beta-blockers (Propranolol)
- Reduces “Tet Spells” by relaxing the right ventricle.
2. Managing “Tet Spells” (Hypercyanotic Episodes)
- Knee-Chest Position (Increases systemic vascular resistance, reducing shunting).
- Administer Oxygen & Morphine (Relaxes pulmonary arteries).
- IV Fluids & Sodium Bicarbonate (Corrects metabolic acidosis).
- Beta-Blockers (Propranolol) to reduce right ventricular outflow obstruction.
Surgical Management:
1. Palliative Surgery (For Severe Cases, Before Full Repair)
- Blalock-Taussig (BT) Shunt:
- Creates a shunt between the subclavian artery & pulmonary artery.
- Temporary measure to increase pulmonary blood flow.
2. Corrective Surgery (Definitive Treatment)
- Total Repair (Complete Intracardiac Repair)
- VSD closure with a synthetic patch.
- Pulmonary stenosis correction (Enlarging the narrowed pulmonary artery or replacing the valve).
- Performed between 3-12 months of age.
- Success rate >95% with improved oxygenation.
Nursing Management:
1. Preoperative Nursing Care:
- Monitor cyanosis & oxygen saturation.
- Teach parents how to manage Tet Spells.
- Administer medications (Propranolol, IV fluids, Oxygen).
- Monitor for signs of dehydration & polycythemia.
- Provide high-calorie nutrition to promote growth.
2. Postoperative Nursing Care:
- Monitor vital signs, oxygen saturation, and signs of heart failure.
- Assess for arrhythmias & bleeding.
- Pain management (IV analgesics).
- Prevent infection (Hand hygiene, aseptic wound care).
- Monitor for signs of pulmonary edema or low cardiac output.
- Encourage early mobilization & deep breathing exercises.
- Parental education on long-term cardiac care.
Complications of TOF:
- Heart Failure (CHF)
- Brain Abscess (Due to prolonged hypoxia)
- Cerebral Thrombosis (Stroke)
- Endocarditis (Infection of heart lining)
- Arrhythmias (After surgery)
- Pulmonary Valve Regurgitation (Post-surgery complication)
Prognosis:
- Untreated TOF → Life expectancy is 2-3 decades.
- Surgical correction (Total Repair) has >95% survival rate.
- Most children lead normal lives after surgery with follow-up care.
Key Points:
✔ TOF is the most common cyanotic congenital heart disease.
✔ Cyanosis, Tet Spells, and the “Boot-shaped heart” are key features.
✔ Beta-blockers & knee-chest positioning help manage Tet Spells.
✔ Surgical correction (Total Repair) is the definitive treatment.
✔ Long-term follow-up ensures good quality of life.
Rheumatic Fever (RF)
Definition:
Rheumatic fever (RF) is an autoimmune inflammatory disease that develops as a complication of untreated or inadequately treated Group A Streptococcal (GAS) pharyngitis (strep throat). It primarily affects children aged 5-15 years and can lead to rheumatic heart disease (RHD), causing permanent damage to heart valves.
Etiology (Causes & Risk Factors):
1. Infectious Cause:
- Group A Beta-Hemolytic Streptococcus (GABHS) infection of the throat.
- Occurs 2-4 weeks after untreated or partially treated strep throat.
2. Immune-Mediated Reaction:
- Molecular mimicry: The immune system mistakenly attacks the body’s tissues (heart, joints, skin, brain) due to similarities between streptococcal proteins and human tissue.
3. Risk Factors:
- Age: Most common in children 5-15 years.
- Poor socioeconomic status (Overcrowding, Poor sanitation).
- Frequent untreated throat infections.
- Genetic predisposition (Family history of RF).
- Living in developing countries (Higher prevalence of RF).
Types of Rheumatic Fever:
1. Acute Rheumatic Fever (ARF)
- Occurs 2-4 weeks after a streptococcal throat infection.
- Self-limiting, but can cause severe cardiac complications.
- Diagnosed based on the modified Jones Criteria.
2. Recurrent Rheumatic Fever
- Occurs in individuals with a previous history of RF.
- Repeated episodes increase the risk of Rheumatic Heart Disease (RHD).
3. Rheumatic Heart Disease (RHD)
- Chronic condition resulting from repeated RF episodes.
- Causes permanent valvular damage (especially the mitral valve).
- Leads to heart failure, atrial fibrillation, embolism.
Pathophysiology:
- Initial Streptococcal Pharyngitis (Strep Throat):
- Group A Streptococcus (GAS) infects the throat.
- The body produces antibodies to fight the infection.
- Autoimmune Cross-Reaction (Molecular Mimicry):
- The immune system mistakes heart, joints, brain, and skin tissues for GAS.
- Inflammatory reaction in multiple organ systems.
- Tissue Damage in Target Organs:
- Heart (Carditis): Valvular damage, endocarditis, myocarditis.
- Joints (Polyarthritis): Inflammation of multiple joints.
- Skin (Subcutaneous nodules, erythema marginatum).
- Brain (Sydenham’s Chorea): Affects the basal ganglia, causing jerky movements.
- Progression to Chronic Rheumatic Heart Disease (RHD)
- Repeated episodes → Fibrosis & scarring of heart valves.
- Mitral valve most commonly affected (Mitral Stenosis, Mitral Regurgitation).
- Leads to heart failure, arrhythmias, stroke.
Clinical Manifestations (Signs & Symptoms):
Major Criteria (Modified Jones Criteria)
- Carditis (Pancarditis)
- Affects endocardium, myocardium, pericardium.
- New heart murmur (Mitral regurgitation, Aortic regurgitation).
- Pericardial friction rub (Pericarditis).
- Signs of heart failure (Dyspnea, Edema, Fatigue).
- Migratory Polyarthritis
- Inflammation of multiple large joints (Knees, Ankles, Elbows, Wrists).
- Pain shifts from one joint to another.
- Redness, warmth, swelling.
- Sydenham’s Chorea (St. Vitus Dance)
- Involuntary, jerky movements of face, hands, and feet.
- Emotional instability, irritability, poor coordination.
- Resolves in weeks to months.
- Erythema Marginatum
- Pink-red, non-itchy rash with wavy margins.
- Appears on trunk & proximal limbs.
- Subcutaneous Nodules
- Painless, firm nodules under the skin.
- Located over bony prominences (Elbows, Knees, Spine).
Minor Criteria:
- Fever (>38.5°C or 101°F).
- Elevated Inflammatory Markers (ESR, CRP).
- Prolonged PR Interval on ECG.
- Arthralgia (Joint pain without swelling).
Supportive Evidence of Recent Strep Infection:
- Positive Throat Culture for Group A Streptococcus.
- Elevated ASO (Anti-Streptolysin O) Titer.
- Positive Rapid Antigen Detection Test (RADT) for GAS.
Diagnosis (Modified Jones Criteria)
Diagnosis is confirmed if:
- 2 Major Criteria OR 1 Major + 2 Minor Criteria are present.
- Evidence of a recent Group A Streptococcal infection.
Imaging & Laboratory Tests:
- Echocardiography (ECHO)
- Detects valvular lesions, chamber enlargement, cardiac dysfunction.
- Electrocardiogram (ECG)
- Prolonged PR Interval (Heart block).
- Chest X-ray
- Cardiomegaly (Enlarged heart).
- Blood Tests
- Elevated ASO Titer, ESR, CRP (Indicate recent streptococcal infection & inflammation).
Medical Management:
1. Antibiotic Therapy (For GAS Eradication)
- Penicillin V (10 days)
- Alternatives: Amoxicillin, Cephalosporins, Erythromycin (If allergic to penicillin).
- Benzathine Penicillin G (IM injection) for prophylaxis.
2. Anti-Inflammatory Treatment
- Aspirin (High Dose)
- For arthritis & carditis (Only in RF, not in viral infections).
- Corticosteroids (Prednisolone)
- For severe carditis & pericarditis.
3. Symptomatic Treatment
- Diuretics (Furosemide) & ACE Inhibitors (Enalapril) for heart failure.
- Beta-Blockers (Propranolol) for Sydenham’s Chorea.
- Haloperidol for severe chorea.
4. Rheumatic Fever Prophylaxis
- Long-term Benzathine Penicillin IM injections to prevent recurrence.
- Duration of Prophylaxis:
- No Carditis → 5 years or until 21 years old.
- Mild Carditis → 10 years or until 21 years old.
- Severe Carditis → Lifetime prophylaxis.
Surgical Management:
- For Chronic Rheumatic Heart Disease (RHD) with Severe Valvular Damage
- Balloon Valvuloplasty – To open narrowed mitral or aortic valves.
- Valve Repair or Replacement – Using prosthetic or biological valves.
- Pacemaker Implantation – If severe arrhythmias develop.
Nursing Management:
1. Acute Phase (Hospitalized Patients)
- Monitor vital signs & cardiac function.
- Assess for heart murmurs, arrhythmias, heart failure symptoms.
- Administer antibiotics, anti-inflammatory medications.
- Provide bed rest during the acute inflammatory phase.
- Apply warm compresses for joint pain relief.
- Monitor neurological status for Sydenham’s Chorea.
2. Discharge Planning & Patient Education
- Importance of completing the full course of antibiotics.
- Prophylactic penicillin injections to prevent recurrence.
- Regular follow-ups & echocardiograms for heart function.
- Good dental hygiene (To prevent infective endocarditis).
- Avoid overcrowded environments & untreated sore throats.
- Encourage vaccination (Influenza, Pneumococcal vaccines).
Complications of Rheumatic Fever:
- Rheumatic Heart Disease (Valvular Stenosis/Regurgitation).
- Congestive Heart Failure.
- Atrial Fibrillation & Thromboembolism.
- Bacterial Endocarditis.
- Stroke (Due to emboli from damaged valves).
Prognosis:
- Acute RF is treatable with antibiotics & anti-inflammatory therapy.
- Untreated RF can lead to lifelong Rheumatic Heart Disease (RHD).
- Early prophylaxis prevents recurrence & cardiac complications.
Key Points:
✔ RF is an autoimmune complication of untreated Strep Throat.
✔ Jones Criteria is used for diagnosis.
✔ Antibiotic prophylaxis prevents recurrence.
✔ RHD can lead to lifelong heart valve damage.
✔ Early intervention prevents complications & improves outcomes.
Rheumatic Heart Disease (RHD)
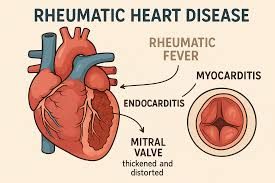
Definition:
Rheumatic Heart Disease (RHD) is a chronic, progressive cardiac condition caused by repeated or severe episodes of rheumatic fever (RF). It leads to permanent damage of the heart valves, mainly the mitral and aortic valves, due to autoimmune inflammation and fibrosis.
Etiology (Causes & Risk Factors):
1. Primary Cause:
- Rheumatic Fever (RF) caused by untreated or inadequately treated Group A Beta-Hemolytic Streptococcus (GABHS) pharyngitis (strep throat).
2. Autoimmune Mechanism:
- Molecular mimicry: The immune system produces antibodies against streptococcal bacteria, which mistakenly attack heart tissue, especially heart valves.
3. Risk Factors:
- Recurrent streptococcal throat infections.
- Poor socioeconomic conditions (Overcrowding, Poor sanitation).
- Genetic predisposition.
- History of untreated or inadequately treated Rheumatic Fever.
- Developing countries (Higher incidence of RHD).
Types of RHD (Based on Valve Involvement):
- Mitral Valve Disease (Most Common – 70-80%)
- Mitral Stenosis: Narrowing of the mitral valve.
- Mitral Regurgitation: Blood flows backward into the left atrium.
- Aortic Valve Disease (Second Most Common – 20-30%)
- Aortic Stenosis: Narrowing of the aortic valve.
- Aortic Regurgitation: Backflow of blood into the left ventricle.
- Tricuspid & Pulmonary Valve Disease (Rare – <5%)
- Usually occurs along with mitral/aortic involvement.
Pathophysiology:
- Rheumatic fever triggers an autoimmune reaction in the heart.
- Chronic inflammation of the heart valves leads to:
- Fibrosis, thickening, and calcification of valve leaflets.
- Valve stenosis (narrowing), causing obstruction to blood flow.
- Valve regurgitation (leakage), leading to volume overload.
- Hemodynamic Consequences:
- Increased workload on the heart → Ventricular hypertrophy.
- Pulmonary congestion (if mitral valve is involved).
- Heart failure due to progressive valve dysfunction.
- Increased risk of atrial fibrillation, thromboembolism, and stroke.
Clinical Manifestations (Signs & Symptoms):
Mild to Moderate RHD (Early Stage)
- Asymptomatic for years.
- Fatigue, weakness.
- Mild dyspnea (Shortness of breath) on exertion.
- Heart murmur detected during routine examination.
Severe RHD (Advanced Disease)
- Exertional dyspnea (Shortness of breath with mild activity).
- Orthopnea (Breathing difficulty while lying flat).
- Paroxysmal Nocturnal Dyspnea (Waking up breathless at night).
- Palpitations (Due to atrial fibrillation).
- Angina (Chest pain, especially with aortic stenosis).
- Edema (Swelling of feet and ankles).
- Ascites (Fluid buildup in the abdomen, if right heart failure develops).
- Cough with frothy sputum (If pulmonary congestion is present).
Complications of RHD
- Heart Failure (Left or right ventricular failure).
- Atrial Fibrillation (AF) (Due to dilated left atrium in mitral stenosis).
- Stroke (Embolism from AF or valve vegetations).
- Pulmonary Hypertension (From chronic mitral valve disease).
- Infective Endocarditis (Bacterial infection of damaged heart valves).
- Sudden Cardiac Death (Severe aortic stenosis can cause fatal arrhythmias).
Diagnosis:
1. Clinical Examination:
- Auscultation Findings:
- Mitral Stenosis: Loud S1, opening snap, diastolic rumbling murmur (Apex).
- Mitral Regurgitation: Pansystolic murmur radiating to the axilla.
- Aortic Stenosis: Harsh systolic ejection murmur (Right 2nd intercostal space).
- Aortic Regurgitation: Diastolic decrescendo murmur (Left sternal border).
2. Imaging & Laboratory Tests:
- Echocardiography (Gold Standard)
- Detects valve stenosis, regurgitation, chamber dilation, pulmonary hypertension.
- Chest X-ray
- Cardiomegaly (Enlarged heart).
- Pulmonary congestion or edema.
- Electrocardiogram (ECG)
- Atrial fibrillation (Irregular P waves).
- Left atrial enlargement in mitral stenosis.
- Right ventricular hypertrophy (If pulmonary hypertension develops).
- Cardiac MRI/CT Scan
- Used in complex cases for detailed valve assessment.
- Doppler Ultrasound (For thromboembolism assessment).
- Blood Tests
- Elevated ESR, CRP (If active inflammation is present).
- Antistreptolysin O (ASO) Titer (Confirms past streptococcal infection).
Medical Management:
1. Prevention of Recurrence
- Long-term Penicillin Prophylaxis:
- Benzathine Penicillin G IM every 3-4 weeks.
- Duration depends on severity:
- No carditis → 5 years or until 21 years old.
- Mild carditis → 10 years or until 21 years old.
- Severe carditis → Lifetime prophylaxis.
2. Symptomatic Treatment
- Diuretics (Furosemide, Spironolactone)
- Reduces pulmonary congestion & edema.
- ACE Inhibitors (Enalapril, Ramipril)
- Reduces afterload & prevents heart failure progression.
- Beta-Blockers (Atenolol, Metoprolol)
- For atrial fibrillation, reduces heart rate & oxygen demand.
- Anticoagulants (Warfarin)
- Prevents stroke in atrial fibrillation cases.
- Digoxin
- Improves heart contractility in severe heart failure.
Surgical Management:
1. Balloon Valvuloplasty (For Mitral or Aortic Stenosis)
- Minimally invasive procedure.
- Uses a balloon catheter to open a narrowed valve.
2. Valve Repair Surgery
- Preferred if native valve can be preserved.
- Common in mitral regurgitation.
3. Valve Replacement Surgery (For Severe Valve Disease)
- Mechanical Valve (Requires lifelong anticoagulation).
- Bioprosthetic Valve (No anticoagulation, but limited lifespan).
- Indicated in patients with severe mitral/aortic stenosis or regurgitation.
4. Heart Transplant (For End-Stage RHD)
- Rarely required, but an option for severe, non-repairable cases.
Nursing Management:
1. Preoperative Nursing Care:
- Monitor respiratory function & signs of heart failure.
- Administer prescribed medications (Diuretics, Beta-blockers, Anticoagulants).
- Educate patients about prophylactic antibiotic therapy.
- Encourage rest & energy conservation.
2. Postoperative Nursing Care (After Valve Surgery)
- Monitor vital signs, ECG, and signs of bleeding.
- Pain management (IV analgesics).
- Encourage early mobilization to prevent thromboembolism.
- Monitor INR (For patients on Warfarin therapy).
- Educate on diet (Avoid vitamin K-rich foods if on Warfarin).
- Ensure regular follow-ups with a cardiologist.
Prognosis:
- Early treatment & prophylaxis prevent RHD progression.
- Surgical correction significantly improves quality of life.
- Without intervention, RHD leads to heart failure & premature death.
Key Points:
✔ RHD is a preventable but serious complication of rheumatic fever.
✔ Mitral & aortic valves are most commonly affected.
✔ Long-term penicillin prophylaxis prevents recurrence.
✔ Surgical valve repair or replacement is necessary for severe cases.
✔ Nursing care focuses on prevention, symptom management, and post-op recovery.
Congestive Cardiac Failure (CCF) / Heart Failure
Definition:
Congestive Cardiac Failure (CCF), or Heart Failure (HF), is a chronic, progressive condition in which the heart is unable to pump sufficient blood to meet the body’s demands. This results in fluid accumulation (congestion) in the lungs and peripheral tissues, leading to symptoms such as shortness of breath, fatigue, and edema.
Etiology (Causes & Risk Factors):
1. Primary Causes of Heart Failure
A. Cardiac Causes:
- Coronary Artery Disease (CAD) – Most common cause.
- Hypertension (HTN) – Increases the workload of the heart.
- Myocardial Infarction (Heart Attack) – Damages heart muscle.
- Valvular Heart Disease (Mitral/Aortic Stenosis, Regurgitation).
- Rheumatic Heart Disease (RHD).
- Congenital Heart Disease (CHD) – ASD, VSD, TOF, PDA.
- Arrhythmias (Atrial fibrillation, Tachycardia-induced cardiomyopathy).
B. Non-Cardiac Causes:
- Chronic Lung Diseases (COPD, Pulmonary Hypertension).
- Anemia (Increases cardiac workload).
- Diabetes Mellitus (Leads to diabetic cardiomyopathy).
- Thyroid Disorders (Hyperthyroidism or Hypothyroidism).
- Chronic Kidney Disease (CKD) (Fluid overload, HTN).
- Alcohol & Drug Abuse (Cocaine, Chemotherapy drugs).
Types of Heart Failure:
1. Based on Ejection Fraction (EF)
- Heart Failure with Reduced Ejection Fraction (HFrEF):
- EF < 40%.
- Caused by systolic dysfunction (weak contraction).
- Heart Failure with Preserved Ejection Fraction (HFpEF):
- EF ≥ 50%.
- Due to diastolic dysfunction (impaired filling).
2. Based on Affected Side
- Left-Sided Heart Failure:
- Failure of the left ventricle → Pulmonary congestion.
- Symptoms: Dyspnea, pulmonary edema, cough, orthopnea.
- Right-Sided Heart Failure:
- Failure of the right ventricle → Peripheral congestion.
- Symptoms: Leg edema, ascites, hepatomegaly, JVD.
- Biventricular Heart Failure:
- Both sides of the heart fail → Combined symptoms.
Pathophysiology:
- Initial Cardiac Insult (MI, HTN, Valvular Disease).
- Decreased Cardiac Output:
- Left-sided failure → Decreased systemic perfusion → Hypoxia.
- Right-sided failure → Blood backs up in venous circulation.
- Compensatory Mechanisms (Temporary Compensation, but Worsens HF):
- Sympathetic Nervous System Activation → Increases HR & BP.
- Renin-Angiotensin-Aldosterone System (RAAS) → Fluid retention.
- Ventricular Hypertrophy (Heart muscle thickens, then weakens).
- Progression to Decompensated Heart Failure:
- Increased fluid overload → Pulmonary & peripheral congestion.
- Decreased organ perfusion → Multi-organ dysfunction (Kidney, Liver, Brain).
Clinical Manifestations (Signs & Symptoms):
1. Left-Sided Heart Failure (Pulmonary Congestion)
- Dyspnea (Shortness of breath) on exertion.
- Orthopnea (Difficulty breathing when lying down).
- Paroxysmal Nocturnal Dyspnea (PND) – Waking up breathless at night.
- Pulmonary Edema (Pink frothy sputum, crackles on auscultation).
- Fatigue, weakness.
- Tachycardia (Compensatory response).
- Cough with blood-tinged sputum.
- Cold, clammy skin (Due to reduced cardiac output).
2. Right-Sided Heart Failure (Peripheral Congestion)
- Jugular Venous Distension (JVD).
- Peripheral Edema (Swelling in legs, feet, hands).
- Ascites (Fluid accumulation in the abdomen).
- Hepatomegaly (Enlarged liver) → Right Upper Quadrant Pain.
- Weight gain due to fluid retention.
- Anorexia & nausea (Due to liver congestion).
Diagnosis:
1. Clinical Examination:
- Auscultation Findings:
- S3 Gallop (Volume overload).
- Bibasilar crackles (Pulmonary congestion).
- Murmurs (If valvular disease is present).
- Peripheral edema, hepatomegaly, JVD assessment.
2. Laboratory Tests:
- B-type Natriuretic Peptide (BNP) > 100 pg/mL → Confirms heart failure.
- N-terminal proBNP (NT-proBNP) > 300 pg/mL → Indicates fluid overload.
- Renal Function Tests (Increased BUN, Creatinine if renal hypoperfusion).
- Liver Function Tests (Elevated ALT, AST due to hepatic congestion).
- Complete Blood Count (CBC) – Anemia can worsen HF.
3. Imaging Studies:
- Echocardiography (Gold Standard):
- Measures Ejection Fraction (EF).
- Identifies ventricular dysfunction, valvular abnormalities.
- Chest X-ray:
- Shows cardiomegaly, pulmonary congestion, pleural effusion.
- Electrocardiogram (ECG):
- Detects arrhythmias, ischemia, LVH.
- Cardiac MRI/CT:
- Used for complex heart failure evaluation.
Medical Management:
1. Lifestyle Modifications
- Salt restriction (<2g/day).
- Fluid restriction (<1.5L/day in severe cases).
- Weight monitoring (Daily weighing to detect fluid retention).
- Regular physical activity (Mild exercise to improve heart function).
2. Pharmacological Therapy
- Diuretics (Furosemide, Spironolactone)
- Reduces pulmonary & peripheral congestion.
- ACE Inhibitors (Enalapril, Ramipril) / ARBs (Losartan)
- Reduces afterload & prevents remodeling.
- Beta-Blockers (Carvedilol, Metoprolol)
- Decreases heart rate & oxygen demand.
- Aldosterone Antagonists (Spironolactone, Eplerenone)
- Reduces fluid overload & potassium loss.
- Digoxin
- Improves contractility in severe HF.
- Anticoagulants (Warfarin)
- For patients with atrial fibrillation to prevent stroke.
Surgical Management:
1. Cardiac Resynchronization Therapy (CRT)
- For patients with left bundle branch block (LBBB).
- Improves ventricular coordination & function.
2. Implantable Cardioverter-Defibrillator (ICD)
- Prevents sudden cardiac death due to arrhythmias.
3. Valve Replacement Surgery
- For heart failure due to valvular disease (Mitral/Aortic valve replacement).
4. Coronary Artery Bypass Graft (CABG)
- For ischemic heart failure due to CAD.
5. Left Ventricular Assist Device (LVAD)
- Used as a bridge to heart transplantation.
6. Heart Transplantation
- For end-stage heart failure not responding to medical therapy.
Nursing Management:
1. Acute Care
- Monitor vital signs, oxygen saturation, lung sounds.
- Administer prescribed medications (Diuretics, ACE inhibitors, Beta-blockers).
- Position patient in High Fowler’s to improve breathing.
- Monitor fluid balance (Intake & Output).
- Monitor for signs of worsening heart failure (Weight gain, dyspnea, edema).
2. Discharge Education
- Medication adherence.
- Low-sodium diet.
- Fluid restriction.
- Daily weight monitoring.
- Recognizing early symptoms of worsening HF.
Prognosis:
- Mild HF: Good prognosis with lifestyle changes & medication.
- Severe HF: Requires long-term management & possible surgical interventions.
- End-stage HF: Consideration for heart transplant.
Key Points:
✔ CCF is a chronic condition with progressive symptoms.
✔ Left-sided HF causes pulmonary congestion, Right-sided HF causes peripheral congestion.
✔ Echocardiography is the gold standard for diagnosis.
✔ Management includes diuretics, ACE inhibitors, beta-blockers, and lifestyle changes.
✔ Advanced cases may require surgical intervention.
Hemophilia

Definition:
Hemophilia is a genetic bleeding disorder in which the blood does not clot properly due to a deficiency of clotting factors VIII or IX. This leads to prolonged bleeding, spontaneous bleeding, and difficulty in stopping hemorrhages.
Etiology (Causes & Risk Factors):
1. Genetic Inheritance:
- X-linked recessive disorder:
- Males are affected (One defective X chromosome).
- Females are usually carriers (Have one normal and one defective X chromosome).
- Spontaneous mutation (30% of cases) can occur in families with no prior history.
2. Deficiency of Clotting Factors:
- Hemophilia A (Classic Hemophilia)
- Deficiency of Factor VIII (8)
- Most common type (80-85%).
- Hemophilia B (Christmas Disease)
- Deficiency of Factor IX (9).
- Less common (15-20%).
- Hemophilia C (Factor XI Deficiency)
- Autosomal recessive disorder (not X-linked).
- Milder bleeding tendency.
- More common in Ashkenazi Jewish populations.
Pathophysiology:
- Clotting cascade disruption:
- Factor VIII or IX deficiency impairs thrombin generation.
- Defective fibrin clot formation:
- Platelets form a weak platelet plug, but fibrin cannot stabilize it.
- Prolonged bleeding:
- Even minor injuries can cause uncontrolled bleeding.
- Recurrent joint and muscle bleeds (Hemarthrosis):
- Bleeding into joints (knees, elbows, ankles) causes pain, swelling, and joint destruction.
Clinical Manifestations (Signs & Symptoms):
Mild Hemophilia (Factor levels 5-40%)
- Prolonged bleeding after surgery, trauma, or dental procedures.
- Rare spontaneous bleeding episodes.
Moderate Hemophilia (Factor levels 1-5%)
- Frequent bruising.
- Prolonged bleeding after minor injuries.
- Occasional spontaneous joint bleeding.
Severe Hemophilia (Factor levels <1%)
- Spontaneous bleeding into joints, muscles, and soft tissues.
- Frequent Hemarthrosis (Joint bleeding) → Joint swelling, pain, restricted movement.
- Muscle Hematomas → Can compress nerves (compartment syndrome).
- Intracranial Bleeding → Can be life-threatening.
- Gastrointestinal (GI) Bleeding → Blood in stool or vomit.
- Hematuria (Blood in urine).
Diagnosis:
1. Laboratory Tests:
- Prothrombin Time (PT) → Normal.
- Activated Partial Thromboplastin Time (aPTT) → Prolonged.
- Clotting Factor Assay → Measures Factor VIII & IX levels.
- Genetic Testing → Identifies carrier status & mutations.
2. Imaging Studies (For Bleeding Complications):
- X-ray/MRI (Joint Bleeding) → Detects joint damage.
- CT/MRI (Head Bleeds) → Detects intracranial hemorrhage.
Medical Management:
1. Clotting Factor Replacement Therapy (Mainstay of Treatment)
- Factor VIII Concentrate (For Hemophilia A).
- Factor IX Concentrate (For Hemophilia B).
- Types of Therapy:
- On-Demand Therapy → Given only during bleeding episodes.
- Prophylactic Therapy → Given regularly to prevent spontaneous bleeding.
2. Desmopressin (DDAVP)
- For Mild Hemophilia A.
- Increases Factor VIII levels.
- Not effective for Hemophilia B.
3. Antifibrinolytic Agents
- Tranexamic Acid, Aminocaproic Acid.
- Prevents clot breakdown (Used for dental procedures, minor bleeds).
4. Pain Management
- Acetaminophen (Paracetamol) → Preferred for joint pain.
- Avoid NSAIDs (Aspirin, Ibuprofen, Naproxen) → Worsen bleeding.
5. Gene Therapy (Experimental)
- Future treatment option to correct defective clotting factor genes.
Surgical Management:
1. Synovectomy
- For recurrent hemarthrosis (Joint bleeding).
- Removes inflamed joint lining to prevent further damage.
2. Joint Replacement Surgery
- For severe joint destruction (Hemophilic arthritis).
- Commonly performed for knees, elbows, ankles.
3. Neurosurgical Intervention
- For intracranial hemorrhage.
Nursing Management:
1. Assessment:
- Monitor for signs of bleeding:
- Joint pain/swelling (Hemarthrosis).
- Sudden severe headache (Intracranial bleeding).
- Blood in urine or stool (Renal/GI bleeding).
- Check vital signs regularly (Tachycardia, hypotension = hypovolemia).
- Assess neurological status (For signs of brain hemorrhage).
- Evaluate pain and joint mobility.
2. Nursing Interventions:
A. Prevent & Manage Bleeding
- Administer clotting factor replacement as prescribed.
- Apply ice packs & immobilization for joint bleeds.
- Encourage use of protective gear (Knee pads, helmets).
- Avoid IM injections & invasive procedures.
- Use soft toothbrushes & electric razors.
B. Pain Management
- Use Acetaminophen (Paracetamol) for pain.
- Avoid NSAIDs & Aspirin.
- Apply warm compresses for muscle hematomas.
C. Prevent Joint Damage
- Encourage gentle range-of-motion exercises after bleeding resolves.
- Avoid weight-bearing activities during acute joint bleeding.
- Use splints/braces to prevent deformities.
D. Education for Patients & Families
- Teach self-administration of factor replacement therapy.
- Recognizing early signs of bleeding (Joint pain, swelling, bruising).
- Importance of regular check-ups & genetic counseling.
- Avoid activities with high risk of trauma (Football, wrestling).
- Inform all healthcare providers about hemophilia status before surgeries or procedures.
Complications of Hemophilia:
- Recurrent Joint Bleeding (Hemarthrosis)
- Leads to chronic joint pain & disability (Hemophilic Arthropathy).
- Intracranial Hemorrhage
- Can cause seizures, paralysis, coma, or death.
- Muscle Hematomas
- Can compress nerves, leading to neuropathy.
- Excessive Postoperative Bleeding
- Requires pre-surgical clotting factor infusion.
- Development of Factor Inhibitors
- Immune system attacks factor concentrates, reducing treatment effectiveness.
Prognosis:
- Mild to Moderate Hemophilia: Good prognosis with proper treatment.
- Severe Hemophilia: High risk of complications, but lifespan can be normal with effective therapy.
- Gene therapy research is ongoing, with potential for permanent cure.
Key Points:
✔ Hemophilia is an X-linked bleeding disorder caused by Factor VIII or IX deficiency.
✔ Major symptom: Prolonged bleeding, spontaneous joint bleeds (Hemarthrosis).
✔ Treatment: Factor replacement therapy, Desmopressin (For mild cases).
✔ Avoid NSAIDs, Aspirin, IM injections, and trauma-prone activities.
✔ Regular prophylactic therapy prevents complications & improves quality of life.
Thalassemia
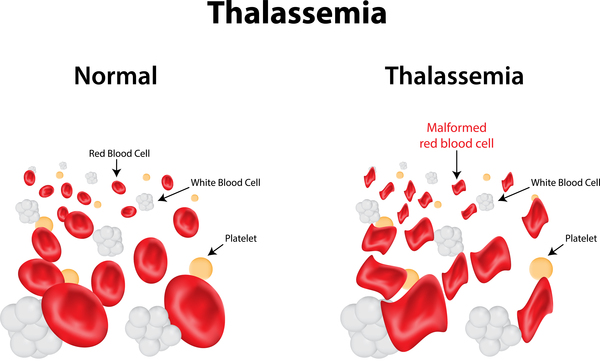
Definition:
Thalassemia is a genetic blood disorder characterized by abnormal hemoglobin production, leading to chronic anemia, hemolysis, and ineffective erythropoiesis. It results from mutations in the genes responsible for producing hemoglobin chains (α-globin or β-globin).
Etiology (Causes & Risk Factors):
1. Genetic Inheritance:
- Autosomal recessive disorder.
- Parents are usually carriers (heterozygous) and pass on the defective gene.
- More common in Mediterranean, South Asian, Middle Eastern, and African populations.
2. Pathogenesis (Defective Hemoglobin Production):
- Hemoglobin (Hb) is made of α & β-globin chains.
- Mutation leads to:
- Reduced or absent globin chain production.
- Unstable hemoglobin that damages red blood cells (RBCs).
- Ineffective erythropoiesis, leading to severe anemia.
Types of Thalassemia:
1. Based on the Affected Globin Chain:
- Alpha (α) Thalassemia → Defective α-globin gene (on chromosome 16).
- Beta (β) Thalassemia → Defective β-globin gene (on chromosome 11).
2. Alpha Thalassemia Subtypes:
| Type | Genetic Defect | Clinical Severity |
|---|---|---|
| Silent Carrier | 1 α-gene deletion | No symptoms |
| Alpha Thalassemia Trait (Minor) | 2 α-gene deletions | Mild anemia |
| Hemoglobin H Disease | 3 α-gene deletions | Moderate-severe anemia |
| Hydrops Fetalis (Hb Bart’s) | 4 α-gene deletions | Fatal in utero |
3. Beta Thalassemia Subtypes:
| Type | Genetic Defect | Clinical Severity |
|---|---|---|
| Beta Thalassemia Minor (Trait) | 1 defective β-gene | Mild anemia |
| Beta Thalassemia Intermedia | Partial reduction in β-globin | Moderate anemia |
| Beta Thalassemia Major (Cooley’s Anemia) | 2 defective β-genes | Severe anemia |
Pathophysiology:
- Defective Hemoglobin Production:
- Reduced or absent α or β chains.
- Unstable hemoglobin forms precipitates inside RBCs.
- Hemolysis & Ineffective Erythropoiesis:
- RBC destruction (hemolysis) in the spleen.
- Bone marrow hyperactivity to compensate → Bone deformities.
- Anemia & Iron Overload:
- Chronic severe anemia → Hypoxia, fatigue, growth retardation.
- Iron overload from blood transfusions → Organ damage (liver, heart, pancreas).
Clinical Manifestations (Signs & Symptoms):
Mild Thalassemia (Silent Carrier, Minor)
- Asymptomatic or mild anemia.
- Mild pallor, fatigue.
Moderate to Severe Thalassemia (Hb H Disease, Intermedia, Major)
- Severe anemia (Hb <7 g/dL).
- Pallor, fatigue, weakness.
- Jaundice (Due to hemolysis).
- Hepatosplenomegaly (Enlarged liver & spleen).
- Skeletal deformities (Frontal bossing, Maxillary overgrowth).
- Growth retardation, Delayed puberty.
- Frequent infections (Due to splenomegaly).
- Iron overload complications (Heart failure, Diabetes, Liver cirrhosis).
Beta Thalassemia Major (Cooley’s Anemia) – Severe Symptoms
- Severe anemia (Transfusion-dependent).
- Characteristic “Chipmunk Face” (Bone marrow expansion).
- Severe splenomegaly (Enlarged spleen requiring removal).
- Cardiac complications (Heart failure due to iron overload).
Diagnosis:
1. Laboratory Tests:
- Complete Blood Count (CBC)
- Low Hemoglobin (Hb) levels.
- Microcytic, hypochromic RBCs (Small, pale RBCs).
- Peripheral Blood Smear
- Target cells, teardrop cells, basophilic stippling.
- Hemoglobin Electrophoresis (Gold Standard)
- Detects abnormal hemoglobin types (Hb A, Hb A2, Hb F, Hb H, Hb Bart’s).
- Iron Studies
- Serum Ferritin ↑ (Iron overload in transfusion-dependent patients).
- Genetic Testing
- Identifies specific α or β-globin gene mutations.
2. Imaging Studies:
- X-ray of Skull & Long Bones
- “Hair-on-end” appearance due to marrow expansion.
- MRI/CT Scan
- Assesses iron overload in heart & liver.
Medical Management:
1. Blood Transfusion Therapy
- For severe anemia (Beta Thalassemia Major, Hb H Disease).
- Regular transfusions (Every 2-4 weeks) maintain Hb >9-10 g/dL.
- Complication: Iron overload (Requires chelation therapy).
2. Iron Chelation Therapy
- To prevent iron overload (Hemosiderosis).
- Drugs used:
- Deferoxamine (IV, SC) – Slow infusion.
- Deferasirox (Oral) – Preferred due to ease of use.
- Deferiprone (Oral) – Alternative for severe cases.
3. Folic Acid Supplementation
- Helps support RBC production.
- Avoid iron supplements (Iron overload risk).
4. Management of Splenomegaly
- Splenectomy (Surgical removal of spleen)
- For patients with severe splenomegaly & transfusion dependence.
- Increases risk of infections → Requires vaccination (Pneumococcal, Meningococcal, H. influenzae).
5. Bone Marrow Transplantation (BMT)
- Only curative treatment.
- Preferred in children with an HLA-matched sibling donor.
- High success rate if performed early.
6. Gene Therapy (Future Treatment)
- Experimental trials focus on correcting the defective globin gene.
Surgical Management:
1. Splenectomy (Spleen Removal)
- Indicated when:
- Excessive splenomegaly causes transfusion dependency.
- Increased RBC destruction leads to severe anemia.
- Post-splenectomy risks:
- Increased risk of infections (Requires lifelong vaccinations).
- Thrombosis risk (Requires prophylactic anticoagulation).
Nursing Management:
1. Assessment
- Monitor for signs of anemia (Pallor, fatigue, weakness).
- Assess for signs of jaundice & splenomegaly.
- Monitor iron overload (Serum ferritin, Cardiac MRI).
- Monitor growth & development in children.
- Assess compliance with transfusion & chelation therapy.
2. Nursing Interventions
A. Managing Anemia & Transfusions
- Ensure timely blood transfusions (Maintain Hb >9-10 g/dL).
- Monitor for transfusion reactions (Fever, chills, rash).
- Encourage hydration to prevent iron overload.
B. Preventing Iron Overload
- Administer iron chelators (Deferoxamine, Deferasirox).
- Monitor liver, heart function (To detect hemosiderosis).
- Educate about iron chelation therapy adherence.
C. Infection Prevention
- Encourage vaccination (Pneumococcal, Meningococcal, H. Influenzae).
- Monitor for fever, signs of infection.
- Educate on post-splenectomy infection risks.
D. Psychological & Growth Support
- Encourage nutritional support (High-protein, vitamin-rich diet).
- Provide emotional support for children with chronic disease.
- Encourage genetic counseling for affected families.
Prognosis:
- Mild cases: Normal life expectancy with minimal intervention.
- Severe cases: Requires lifelong transfusions & iron chelation.
- Bone marrow transplant can cure thalassemia major if performed early.
Key Points:
✔ Thalassemia is an inherited disorder causing defective hemoglobin synthesis.
✔ Beta Thalassemia Major is transfusion-dependent & causes severe anemia.
✔ Iron overload from transfusions requires chelation therapy.
✔ Bone marrow transplantation is the only curative treatment.
✔ Nursing care focuses on transfusion safety, infection prevention, and psychological support.
Anemia
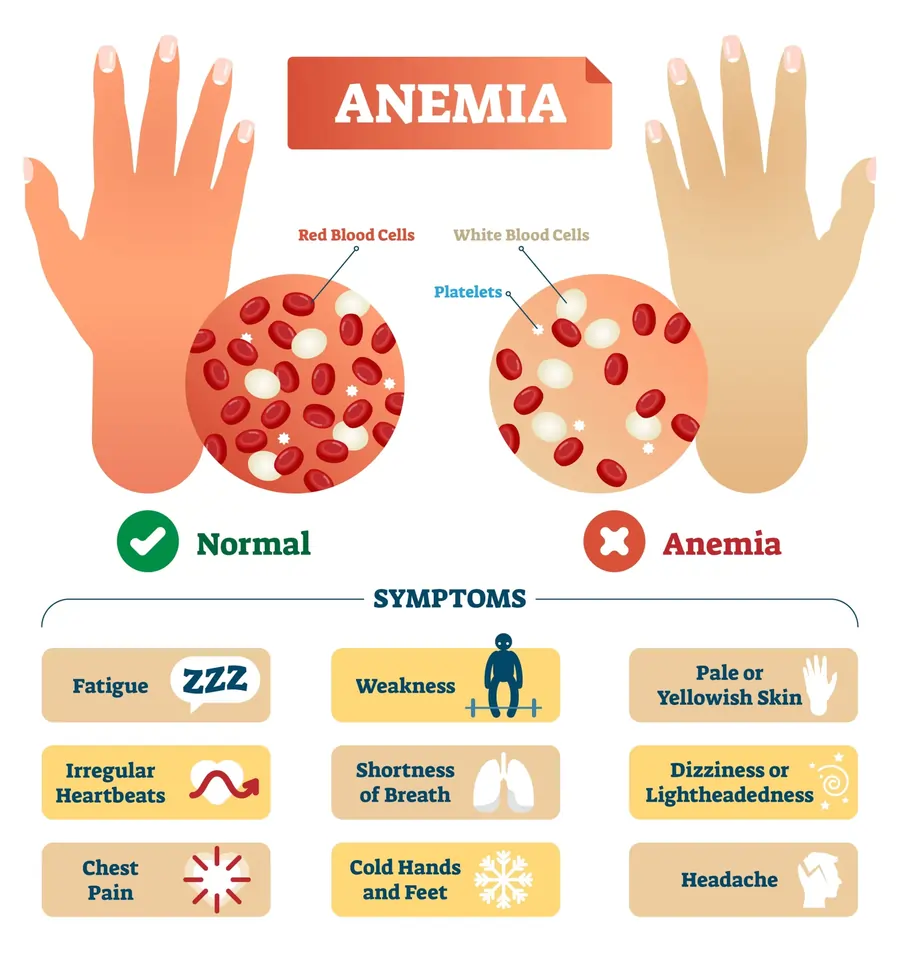
Definition:
Anemia is a hematological disorder characterized by a decrease in the number of red blood cells (RBCs), hemoglobin (Hb), or hematocrit levels, resulting in reduced oxygen-carrying capacity of the blood. This leads to tissue hypoxia and various systemic symptoms.
- Normal Hemoglobin Levels:
- Men: 13.8–17.2 g/dL
- Women: 12.1–15.1 g/dL
- Children: 11–16 g/dL
Etiology (Causes & Risk Factors):
1. Based on Pathophysiology:
- Decreased RBC Production (Bone marrow failure, nutritional deficiency).
- Increased RBC Destruction (Hemolysis) (Sickle cell anemia, Thalassemia, Autoimmune hemolysis).
- Blood Loss (Acute or chronic bleeding).
2. Common Causes of Anemia:
- Nutritional Deficiencies:
- Iron Deficiency (Most Common)
- Vitamin B12 Deficiency (Pernicious Anemia)
- Folic Acid Deficiency
- Chronic Diseases:
- Chronic Kidney Disease (Erythropoietin deficiency)
- Liver Disease (Hemolysis & clotting abnormalities)
- Autoimmune Disorders (Lupus, Rheumatoid Arthritis)
- Bone Marrow Disorders:
- Aplastic Anemia
- Myelodysplastic Syndrome
- Leukemia
- Genetic Disorders:
- Sickle Cell Anemia
- Thalassemia
- Acute or Chronic Blood Loss:
- Gastrointestinal Bleeding (Ulcers, Colon Cancer, Hemorrhoids)
- Menorrhagia (Heavy menstrual bleeding)
- Trauma, Surgery, Postpartum Hemorrhage
- Infections & Toxins:
- Malaria, Parvovirus B19
- Lead Poisoning
Types of Anemia:
1. Based on Morphology (RBC Size & Shape)
| Type | RBC Morphology | Common Causes |
|---|---|---|
| Microcytic Hypochromic Anemia | Small, pale RBCs | Iron deficiency, Thalassemia, Lead poisoning |
| Normocytic Normochromic Anemia | Normal-sized RBCs | Acute blood loss, Chronic disease, Aplastic anemia |
| Macrocytic (Megaloblastic) Anemia | Large RBCs | Vitamin B12 or Folic Acid Deficiency, Alcoholism, Hypothyroidism |
Pathophysiology:
1. Iron Deficiency Anemia (Most Common Type)
- Decreased iron stores → Reduced hemoglobin synthesis → Microcytic, hypochromic RBCs → Tissue hypoxia.
2. Megaloblastic Anemia (Vitamin B12/Folate Deficiency)
- Defective DNA synthesis → Large, fragile RBCs → Ineffective erythropoiesis & premature RBC destruction.
3. Hemolytic Anemia (Increased RBC Destruction)
- Autoimmune reaction or genetic defect → Destruction of RBCs before their normal lifespan → Hemoglobinuria, Jaundice, Splenomegaly.
4. Anemia of Chronic Disease
- Inflammation increases hepcidin → Inhibits iron absorption & release from stores → Mild-moderate anemia.
Clinical Manifestations (Signs & Symptoms):
1. General Symptoms (All Types of Anemia)
- Fatigue, Weakness
- Pallor (Pale skin, conjunctiva, and mucous membranes)
- Shortness of Breath (Dyspnea)
- Dizziness, Headache
- Cold Hands and Feet
- Tachycardia (Increased Heart Rate)
- Orthostatic Hypotension (Dizziness on standing up)
2. Specific Symptoms Based on Type
A. Iron Deficiency Anemia
- Glossitis (Inflamed tongue)
- Koilonychia (Spoon-shaped nails)
- Pica (Craving for non-food items like ice, clay)
B. Vitamin B12 Deficiency (Pernicious Anemia)
- Neurological symptoms (Tingling, Numbness, Memory Loss)
- Beefy red tongue
- Ataxia (Loss of coordination)
C. Hemolytic Anemia
- Jaundice (Yellowing of skin & eyes)
- Dark-colored urine (Hemoglobinuria)
- Splenomegaly (Enlarged spleen)
D. Aplastic Anemia
- Pancytopenia (Low RBCs, WBCs, and Platelets)
- Recurrent infections
- Easy bruising & bleeding (Petechiae, Purpura)
Diagnosis:
1. Laboratory Tests
- Complete Blood Count (CBC):
- Low Hemoglobin & Hematocrit (Hct)
- RBC Indices (MCV, MCH, MCHC) determine anemia type.
- Peripheral Blood Smear:
- Microcytic RBCs (Iron Deficiency, Thalassemia).
- Macrocytic RBCs (B12/Folate Deficiency).
- Sickle Cells, Target Cells (Hemolytic Anemias).
- Serum Ferritin & Iron Studies:
- Low Iron, Low Ferritin → Iron Deficiency.
- High Ferritin, Low Iron → Chronic Disease.
- Reticulocyte Count:
- Increased → Hemolysis or Blood Loss.
- Decreased → Bone Marrow Failure.
- Bone Marrow Biopsy:
- For suspected Aplastic Anemia, Leukemia.
- Vitamin B12 & Folate Levels:
- Low in Megaloblastic Anemia.
- Hemoglobin Electrophoresis:
- For Sickle Cell Anemia & Thalassemia.
- Coombs Test:
- Confirms Autoimmune Hemolytic Anemia.
Medical Management:
1. Iron Deficiency Anemia
- Iron Supplementation (Ferrous Sulfate, Ferrous Gluconate).
- Iron-Rich Diet (Red meat, Spinach, Lentils, Beans).
- IV Iron Therapy (Severe cases, Malabsorption).
2. Vitamin B12 & Folic Acid Deficiency
- Vitamin B12 Injections (Cyanocobalamin IM for Pernicious Anemia).
- Folic Acid Supplements.
- Diet: Green leafy vegetables, Dairy, Eggs, Fish.
3. Hemolytic Anemia
- Corticosteroids (Prednisolone for Autoimmune Hemolysis).
- Blood Transfusions (Severe cases).
- Plasmapheresis (For Autoimmune Hemolysis).
- Splenectomy (For hereditary spherocytosis, severe cases).
4. Aplastic Anemia
- Bone Marrow Transplant (Curative for Severe Cases).
- Immunosuppressive Therapy (Cyclosporine, ATG).
5. Anemia of Chronic Disease
- Erythropoietin Injections (CKD Patients).
- Manage Underlying Condition (Inflammation, Infections, Cancer).
Surgical Management:
- Splenectomy (For Severe Hemolytic Anemia)
- Bone Marrow Transplant (For Aplastic Anemia, Severe Thalassemia)
- Gastrointestinal Surgery (For Chronic GI Bleeding, Peptic Ulcer)
Nursing Management:
1. Assessment
- Monitor Hemoglobin, RBC, and Reticulocyte Count.
- Assess for Symptoms of Hypoxia (Tachycardia, Dyspnea, Confusion).
- Evaluate Nutritional Intake (Iron, B12, Folic Acid).
- Assess for Bleeding (Petechiae, Hematuria, Melena).
2. Interventions
- Administer Iron, Vitamin B12, or Erythropoietin as prescribed.
- Encourage Diet Rich in Essential Nutrients.
- Monitor for Blood Transfusion Reactions.
- Prevent Infection in Immunocompromised Patients.
- Educate on Medication Adherence & Follow-ups.
Prognosis:
- Mild anemia: Treatable with diet & supplements.
- Severe anemia: Requires transfusions, possible surgery.
- Aplastic & Hemolytic Anemia: Lifelong management needed.
Key Points:
✔ Anemia is caused by decreased RBC production, hemolysis, or blood loss.
✔ Iron Deficiency Anemia is the most common type.
✔ Diagnosis includes CBC, Iron Studies, B12/Folate Levels, Bone Marrow Biopsy.
✔ Treatment depends on the cause (Iron, B12, Blood Transfusion, Bone Marrow Transplant).
✔ Nursing care focuses on monitoring symptoms, providing supplements, and preventing complications.
Leukemia in Children
Definition:
Leukemia is a malignant disorder of the bone marrow and blood, characterized by the uncontrolled proliferation of immature white blood cells (WBCs or blasts). These abnormal WBCs crowd the bone marrow, interfering with normal blood cell production, leading to anemia, infections, and bleeding disorders.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Chromosomal abnormalities:
- Down Syndrome (Trisomy 21) increases leukemia risk.
- Philadelphia Chromosome (BCR-ABL fusion gene) in chronic myeloid leukemia (CML).
- Li-Fraumeni Syndrome, Fanconi Anemia (Inherited conditions with increased cancer risk).
2. Environmental Factors:
- Exposure to ionizing radiation (e.g., radiation therapy, nuclear exposure).
- Exposure to chemicals (e.g., benzene, pesticides, chemotherapy drugs).
- Viral infections (e.g., Epstein-Barr Virus, Human T-lymphotropic Virus – HTLV).
3. Immune System Dysfunction:
- Weakened immune system (HIV, post-organ transplant, chemotherapy).
- Autoimmune disorders.
Types of Leukemia in Children:
1. Based on the Affected Cell Lineage:
| Type | Description | Prevalence |
|---|---|---|
| Acute Lymphoblastic Leukemia (ALL) | Overproduction of immature lymphoid precursor cells (Lymphoblasts) | Most common (75-80% of childhood leukemias) |
| Acute Myeloid Leukemia (AML) | Overproduction of immature myeloid precursor cells (Myeloblasts) | 15-20% of childhood leukemias |
| Chronic Myeloid Leukemia (CML) | Slow-growing leukemia with abnormal myeloid WBCs due to Philadelphia Chromosome | Rare in children |
| Chronic Lymphocytic Leukemia (CLL) | Slow-growing leukemia affecting lymphocytes | Extremely rare in children |
2. Based on Progression Rate:
- Acute Leukemia: Rapid onset, severe symptoms, requires urgent treatment.
- Chronic Leukemia: Slow progression, often diagnosed in later stages.
Pathophysiology:
- Mutation in Bone Marrow Stem Cells → Uncontrolled proliferation of immature WBCs (Blasts).
- Bone Marrow Overcrowding → Inhibits the production of normal RBCs, WBCs, and platelets.
- ↓ RBCs → Anemia (Fatigue, Pallor).
- ↓ WBCs → Infections (Fever, Weak Immune Response).
- ↓ Platelets → Bleeding tendencies (Bruising, Petechiae).
- Blast Cells Invade Other Organs:
- Hepatosplenomegaly (Liver & Spleen Enlargement).
- Lymphadenopathy (Swollen Lymph Nodes).
- CNS Infiltration (Headache, Seizures, Vision Changes).
Clinical Manifestations (Signs & Symptoms):
1. General Symptoms:
- Pallor, Fatigue, Weakness (Due to anemia).
- Frequent infections, Fever, Mouth sores (Due to neutropenia).
- Easy bruising, Petechiae, Epistaxis (Nosebleeds), Bleeding gums (Due to thrombocytopenia).
- Bone & Joint Pain (Due to blast cell accumulation in bone marrow).
- Unexplained weight loss, Loss of appetite.
- Swollen lymph nodes (Neck, Armpits, Groin).
2. Specific Symptoms Based on Organ Involvement:
| Organ | Symptoms |
|---|---|
| Liver & Spleen | Hepatosplenomegaly (Enlarged liver & spleen) → Abdominal swelling |
| Central Nervous System (CNS) | Headache, Seizures, Blurred vision, Vomiting |
| Testicles (Boys) | Swelling, Firmness (Common in ALL) |
Diagnosis:
1. Blood Tests:
- Complete Blood Count (CBC)
- ↑ WBCs (Blasts in peripheral blood)
- ↓ RBCs (Anemia)
- ↓ Platelets (Thrombocytopenia)
- Peripheral Blood Smear:
- Blast cells present in blood.
2. Bone Marrow Aspiration & Biopsy (Gold Standard)
- Confirms diagnosis by detecting >20% blasts in bone marrow.
3. Cytogenetic & Molecular Studies:
- Philadelphia Chromosome (CML)
- MLL Gene Rearrangements (High-risk ALL)
4. Lumbar Puncture (Spinal Tap)
- Checks for CNS Involvement (Leukemic cells in cerebrospinal fluid – CSF).
5. Imaging Studies (CT/MRI, X-ray)
- To detect organ enlargement & bone involvement.
Medical Management:
1. Chemotherapy (Primary Treatment)
- Induction Phase: High-dose chemotherapy to destroy leukemic cells.
- Consolidation Phase: Prevents relapse.
- Maintenance Phase: Low-dose chemo for 2-3 years to prevent recurrence.
2. Targeted Therapy
- Tyrosine Kinase Inhibitors (Imatinib, Dasatinib)
- Used in Philadelphia Chromosome-positive (CML) leukemia.
3. Immunotherapy
- CAR-T Cell Therapy (Chimeric Antigen Receptor Therapy)
- Modifies patient’s immune cells to attack leukemia cells.
4. Radiation Therapy
- For CNS involvement or testicular leukemia.
5. Supportive Care
- Antibiotics, Antifungals, Antivirals (To prevent infections).
- Blood Transfusions (For severe anemia & thrombocytopenia).
- Pain Management (Acetaminophen, Morphine for bone pain).
Surgical Management:
1. Bone Marrow Transplant (Stem Cell Transplant)
- For relapsed or high-risk leukemia.
- Requires HLA-matched donor (Sibling preferred).
- High-risk but potentially curative.
2. Splenectomy (Rare)
- For severe splenomegaly causing thrombocytopenia.
Nursing Management:
1. Infection Prevention
- Maintain strict hand hygiene & isolation precautions.
- Administer prophylactic antibiotics & antifungals.
- Avoid live vaccines (MMR, Varicella) in immunosuppressed children.
- Encourage a well-balanced diet to maintain immunity.
2. Managing Chemotherapy Side Effects
- Monitor for nausea, vomiting, diarrhea (Give antiemetics).
- Manage mucositis (Oral care with saline rinses, avoid spicy foods).
- Watch for signs of neutropenia (Fever, sore throat, cough).
- Monitor renal function (Methotrexate toxicity can damage kidneys).
3. Bleeding Precautions
- Monitor for petechiae, epistaxis, gum bleeding.
- Use soft toothbrushes, avoid IM injections & rough play.
- Administer platelet transfusions if needed.
4. Pain & Comfort Management
- Encourage mild physical activity to prevent muscle wasting.
- Administer prescribed analgesics (Avoid NSAIDs due to bleeding risk).
- Provide emotional support to the child & family.
5. Psychosocial Support
- Support coping mechanisms in children & parents.
- Encourage interaction with other children with leukemia.
- Provide education about disease, treatment, and prognosis.
Complications of Leukemia:
- Tumor Lysis Syndrome (Life-threatening electrolyte imbalance).
- Sepsis (Due to neutropenia & immunosuppression).
- Graft-Versus-Host Disease (After bone marrow transplant).
- Organ Damage (Heart, Liver, Kidney due to chemotherapy).
- Relapse (Leukemia returning after remission).
Prognosis:
- ALL (Survival Rate: 85-90%) with early treatment.
- AML (Survival Rate: 50-70%).
- CML (Good prognosis with targeted therapy).
- Early diagnosis & aggressive treatment improve outcomes.
Key Points:
✔ Leukemia is a cancer of WBCs, leading to anemia, infections, and bleeding.
✔ ALL is the most common childhood leukemia, followed by AML.
✔ Diagnosis is confirmed by bone marrow biopsy (>20% blasts).
✔ Treatment includes chemotherapy, targeted therapy, stem cell transplant.
✔ Nursing care focuses on infection prevention, chemotherapy management, and psychosocial support.
Hodgkin’s and Non-Hodgkin’s Lymphoma in Children
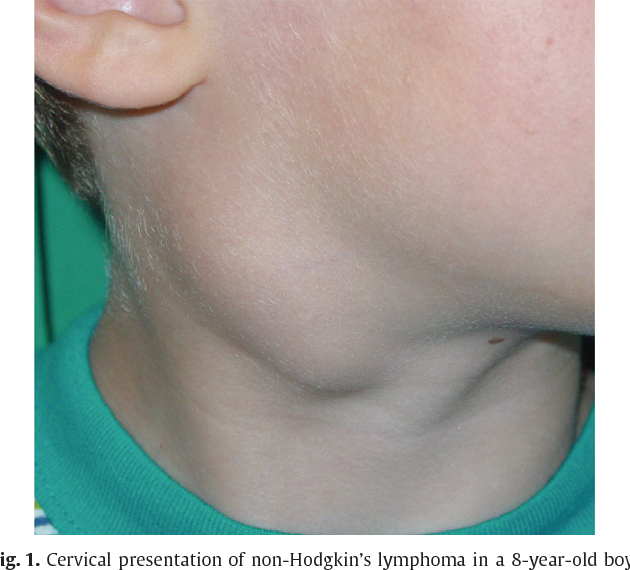
Definition:
Lymphomas are malignant cancers of the lymphatic system, characterized by uncontrolled proliferation of abnormal lymphocytes in the lymph nodes, spleen, thymus, and other lymphoid tissues.
Types of Lymphoma:
- Hodgkin’s Lymphoma (HL)
- Characterized by the presence of Reed-Sternberg cells.
- Often affects older children & adolescents (ages 10-19).
- Non-Hodgkin’s Lymphoma (NHL)
- No Reed-Sternberg cells, more aggressive.
- More common in younger children.
Etiology (Causes & Risk Factors):
1. Genetic & Environmental Factors
- Weakened immune system (HIV, Organ Transplant, Chemotherapy).
- Epstein-Barr Virus (EBV) association (More common in HL).
- Radiation exposure & exposure to carcinogens.
2. Genetic Mutations & Chromosomal Abnormalities
- Mutations in B-cell or T-cell lymphocytes lead to uncontrolled proliferation.
- Translocations in chromosome 8 (Burkitt’s Lymphoma – NHL).
Pathophysiology:
- Genetic mutation in lymphocytes (B-cells or T-cells).
- Uncontrolled growth of malignant lymphocytes in lymph nodes & tissues.
- Lymph node enlargement & infiltration of nearby organs (Spleen, Liver, Bone Marrow, CNS).
- Systemic symptoms due to excessive cytokine production (Fever, Night Sweats, Weight Loss).
Types of Lymphomas in Children:
1. Hodgkin’s Lymphoma (HL)
- Classical Hodgkin’s Lymphoma (CHL) (Most common type).
- Nodular Lymphocyte-Predominant HL (Rare in children).
2. Non-Hodgkin’s Lymphoma (NHL)
| Type | Cell Origin | Characteristics |
|---|---|---|
| Burkitt’s Lymphoma | B-cell | Highly aggressive, rapid tumor growth |
| Lymphoblastic Lymphoma | T-cell or B-cell | Often involves mediastinum |
| Diffuse Large B-cell Lymphoma (DLBCL) | B-cell | Common in older children |
| Anaplastic Large Cell Lymphoma (ALCL) | T-cell | Rare but aggressive |
Clinical Manifestations (Signs & Symptoms):
1. Hodgkin’s Lymphoma (HL) Symptoms
- Painless, enlarged lymph nodes (Cervical, Supraclavicular, Axillary).
- B Symptoms (Systemic symptoms associated with advanced disease):
- Fever (>38°C).
- Drenching night sweats.
- Unexplained weight loss (>10% in 6 months).
- Fatigue, Itching (Pruritus).
- Splenomegaly, Hepatomegaly (Advanced cases).
2. Non-Hodgkin’s Lymphoma (NHL) Symptoms
- Rapidly growing lymph nodes (Neck, Abdomen, Chest).
- Swelling in the face & neck (Superior Vena Cava Syndrome – Emergency!).
- Abdominal pain, nausea (If abdominal lymph nodes are involved).
- Difficulty breathing, cough (If mediastinal mass present).
- CNS involvement (Seizures, Headache, Confusion).
Diagnosis:
1. Laboratory Tests
- Complete Blood Count (CBC)
- Anemia, leukocytosis, or thrombocytopenia (Bone marrow involvement).
- Erythrocyte Sedimentation Rate (ESR)
- Elevated in Hodgkin’s Lymphoma.
- Lactate Dehydrogenase (LDH) (Elevated in NHL, Indicates rapid tumor growth).
2. Lymph Node Biopsy (Gold Standard)
- Hodgkin’s Lymphoma: Presence of Reed-Sternberg Cells.
- Non-Hodgkin’s Lymphoma: Diffuse malignant lymphocyte proliferation.
3. Imaging Studies:
- Chest X-ray (Mediastinal mass in NHL).
- CT/MRI Scan (Staging, Tumor Spread).
- Positron Emission Tomography (PET) Scan (For metastasis).
4. Bone Marrow Aspiration & Lumbar Puncture
- To detect bone marrow & CNS involvement.
Medical Management:
1. Chemotherapy (Primary Treatment)
- HL Treatment Regimens:
- ABVD (Adriamycin, Bleomycin, Vinblastine, Dacarbazine).
- BEACOPP (For advanced HL).
- NHL Treatment Regimens:
- CHOP (Cyclophosphamide, Doxorubicin, Vincristine, Prednisone).
- Intensive chemotherapy for Burkitt’s Lymphoma.
2. Radiation Therapy
- Used in Hodgkin’s Lymphoma for localized disease.
- Limited use in NHL (More systemic disease).
3. Immunotherapy & Targeted Therapy
- Rituximab (Anti-CD20 for B-cell Lymphomas).
- CAR-T Cell Therapy (For relapsed NHL).
4. Corticosteroids (Dexamethasone, Prednisone)
- Reduces tumor swelling & inflammation.
Surgical Management:
- Lymph Node Biopsy (Diagnostic purpose).
- Surgical Removal of Isolated Tumors (Rare in lymphoma).
Nursing Management:
1. Monitor for Chemotherapy Side Effects
- Bone Marrow Suppression (Neutropenia, Thrombocytopenia, Anemia).
- Manage nausea/vomiting (Give Antiemetics like Ondansetron).
- Monitor for mucositis (Oral care with saline rinses).
2. Infection Prevention
- Neutropenic Precautions (Reverse Isolation, Avoid Crowds).
- Monitor for fever (Early sign of infection in immunocompromised children).
- Administer prophylactic antibiotics if needed.
3. Manage Tumor Lysis Syndrome (TLS)
- Hydration & Electrolyte Monitoring (Risk of hyperkalemia, hyperuricemia).
- Allopurinol (Prevents uric acid buildup from cell breakdown).
4. Prevent Complications
- Superior Vena Cava Syndrome (SVCS) (Swelling in face, airway obstruction).
- Spinal Cord Compression (Paralysis, Severe Back Pain).
5. Psychosocial Support
- Provide emotional support to the child & family.
- Encourage interaction with support groups & other pediatric cancer patients.
- Assist with coping strategies for hair loss & body image changes.
Complications of Lymphoma:
- Bone Marrow Suppression → Increased risk of infections & bleeding.
- Secondary Malignancies (Long-term chemotherapy/radiation effects).
- Infertility (Due to chemotherapy/radiation).
- Neuropathy (From chemotherapy drugs like Vincristine).
Prognosis:
- Hodgkin’s Lymphoma: Excellent prognosis (5-year survival rate >90%).
- Non-Hodgkin’s Lymphoma: Survival rates vary (70-90% depending on type & stage).
- Early diagnosis & aggressive treatment improve outcomes.
Key Points:
✔ Lymphoma is a cancer of the lymphatic system, classified as Hodgkin’s or Non-Hodgkin’s Lymphoma.
✔ HL has Reed-Sternberg cells, NHL does not.
✔ Common symptoms include painless lymph node swelling, fever, night sweats, and weight loss.
✔ Treatment includes chemotherapy, radiation, immunotherapy, and supportive care.
✔ Nursing care focuses on infection prevention, managing chemotherapy side effects, and psychosocial support.
Gastro-intestinal system:
- Identification and Nursing
management of congenital
malformations.
Cleft Lip in Children

Definition:
Cleft lip is a congenital anomaly characterized by a split or opening in the upper lip, resulting from failure of the maxillary and medial nasal processes to fuse properly during fetal development. It can occur unilaterally or bilaterally and may or may not be associated with a cleft palate.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Family history of cleft lip and/or cleft palate.
- Chromosomal abnormalities (e.g., Trisomy 13, Van der Woude syndrome).
2. Environmental Factors:
- Maternal smoking, alcohol consumption, drug abuse.
- Maternal malnutrition (Folic acid, Vitamin B deficiency).
- Use of certain medications during pregnancy (Antiepileptics, Retinoids, Steroids).
- Exposure to teratogens (Radiation, Chemicals, Viral Infections – Rubella, Cytomegalovirus).
Types of Cleft Lip:
| Type | Description |
|---|---|
| Unilateral Cleft Lip | Occurs on one side of the upper lip. |
| Bilateral Cleft Lip | Occurs on both sides of the upper lip. |
| Incomplete Cleft Lip | A partial split in the lip that does not extend into the nostril. |
| Complete Cleft Lip | A full split extending into the nostril, often associated with cleft palate. |
Pathophysiology:
- During the 4th-7th week of embryonic development, the maxillary and medial nasal processes fail to fuse properly.
- This results in a gap or cleft in the upper lip, which may or may not involve the alveolar ridge and palate.
- Cleft lip affects feeding, speech development, and facial aesthetics, and may be associated with other syndromic abnormalities.
Clinical Manifestations (Signs & Symptoms):
1. Visible Facial Deformity
- Unilateral or Bilateral cleft in the upper lip.
- Gap may extend to the nostril (Severe cases).
2. Feeding Difficulties
- Difficulty latching onto the breast or bottle.
- Poor suction and milk regurgitation through the nose (If associated with cleft palate).
3. Speech & Language Issues (If Not Corrected Early)
- Delayed speech development.
- Nasal-sounding speech due to air leakage.
4. Dental & Orthodontic Problems
- Misalignment of teeth.
- Higher risk of dental caries & infections.
5. Ear Infections & Hearing Loss (If Associated with Cleft Palate)
- Increased risk of otitis media (Middle ear infections).
- Hearing impairment due to Eustachian tube dysfunction.
Diagnosis:
1. Prenatal Diagnosis
- Ultrasound (18-22 weeks gestation) – Detects facial abnormalities.
2. Postnatal Diagnosis
- Physical Examination – Clearly visible cleft on the upper lip.
- Feeding Assessment – Evaluates sucking ability.
- Hearing & Speech Evaluation – If cleft palate is involved.
- Genetic Testing – To rule out syndromic associations.
Medical Management:
1. Nutritional Support (Before Surgery)
- Special feeding techniques (Use of specialized bottles like Haberman Feeder).
- Frequent burping to prevent aspiration.
- Encouraging breastfeeding if possible, or using soft bottle nipples.
2. Speech Therapy
- For speech difficulties after surgical repair.
3. Dental & Orthodontic Care
- Early dental evaluation & orthodontic treatment to correct misaligned teeth.
Surgical Management (Definitive Treatment):
1. Timing of Surgery
- Cleft Lip Repair (Cheiloplasty) → Done at 3-6 months of age.
- Cleft Palate Repair (Palatoplasty) → Done at 9-18 months of age.
2. Surgical Techniques for Cleft Lip Repair
- Millard Rotation-Advancement Flap Technique (Most commonly used).
- Tennison-Randall Triangular Flap Technique (For wider clefts).
3. Goals of Surgery
- Close the cleft & restore normal lip function.
- Improve facial symmetry & appearance.
- Enhance speech development & feeding ability.
4. Postoperative Care
- Suture care & wound healing monitoring.
- Preventing infection (Antibiotic ointment, avoiding excessive crying).
- Pain management (Acetaminophen, Ibuprofen).
- Restraints (Elbow splints) to prevent the child from touching the surgical site.
Nursing Management:
1. Preoperative Nursing Care
- Assess feeding difficulties & teach parents alternative feeding methods (Use of specialized bottles, breastfeeding techniques).
- Monitor weight gain & nutritional status.
- Provide emotional support to parents (Addressing concerns about appearance & future speech development).
- Educate parents about surgical repair & postoperative care.
2. Postoperative Nursing Care
A. Pain Management
- Administer prescribed analgesics (Acetaminophen, Ibuprofen).
- Provide comfort measures (Rocking, Cuddling).
B. Preventing Surgical Site Complications
- Keep the surgical site clean & dry.
- Apply antibiotic ointment as prescribed.
- Use elbow restraints to prevent the child from touching the stitches.
- Avoid pacifiers, straws, or hard foods that may disturb the sutures.
C. Feeding & Nutrition
- Encourage breastfeeding if possible (Using a soft nipple bottle if needed).
- Use spoon-feeding or cup-feeding postoperatively (Avoid sucking motion).
- Provide soft, pureed foods until healing is complete.
D. Monitor for Complications
- Watch for signs of infection (Redness, Swelling, Fever).
- Monitor for respiratory distress (Rare but possible due to swelling).
E. Parental Education
- Explain proper wound care & feeding techniques.
- Emphasize follow-up visits with the surgeon, speech therapist, and orthodontist.
- Encourage emotional bonding & psychosocial support.
Complications of Cleft Lip (If Untreated):
- Severe feeding difficulties → Malnutrition & growth retardation.
- Speech impairment → Delayed language development, articulation issues.
- Frequent ear infections → Hearing loss & learning difficulties.
- Dental abnormalities → Misaligned teeth, increased risk of cavities.
- Psychosocial issues → Poor self-esteem, social isolation.
Prognosis:
- With early surgical repair, feeding therapy, and speech therapy, children with cleft lip can lead a normal life.
- Most children have minimal scarring and normal speech development if treated early.
Key Points:
✔ Cleft lip is a congenital facial anomaly due to failure of fusion of the maxillary and nasal processes.
✔ It can be unilateral or bilateral and may or may not involve the palate.
✔ Diagnosis is done via prenatal ultrasound or postnatal physical examination.
✔ **Definitive treatment is surgical repair (Cheiloplasty at 3-6 months).
✔ Postoperative care includes pain management, wound care, and feeding modifications.
✔ Nursing care focuses on preoperative feeding support, postoperative wound care, and family education.
Cleft Palate in Children
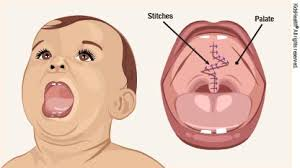
Definition:
Cleft palate is a congenital defect in which there is an opening in the roof of the mouth (palate) due to incomplete fusion of the palatal shelves during fetal development. It can occur alone or in combination with a cleft lip.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Family history of cleft palate or syndromes involving craniofacial abnormalities.
- Chromosomal disorders (Trisomy 13, Pierre Robin Sequence, Van der Woude Syndrome).
2. Environmental Factors:
- Maternal smoking, alcohol consumption, or drug use during pregnancy.
- Maternal folic acid deficiency.
- Exposure to teratogens (Radiation, Pesticides, Chemicals).
- Certain medications during pregnancy (Antiepileptics, Retinoids, Corticosteroids).
Types of Cleft Palate:
| Type | Description |
|---|---|
| Incomplete Cleft Palate | Partial opening in the soft or hard palate. |
| Complete Cleft Palate | Extends through the entire soft & hard palate, may involve the uvula. |
| Unilateral Cleft Palate | Occurs on one side of the palate. |
| Bilateral Cleft Palate | Occurs on both sides, often associated with cleft lip. |
| Submucous Cleft Palate | Hidden cleft where the mucous membrane is intact, but muscles underneath are split. |
Pathophysiology:
- During weeks 7-12 of fetal development, the palatal shelves fail to fuse properly, leaving an opening in the roof of the mouth.
- This results in:
- Communication between the oral and nasal cavities.
- Difficulty in feeding (milk escapes into the nasal cavity).
- Speech difficulties (Air escapes through the nose).
- Increased risk of ear infections (Middle ear dysfunction due to Eustachian tube abnormalities).
Clinical Manifestations (Signs & Symptoms):
1. Visible Deformity
- Opening in the hard/soft palate, sometimes extending into the nasal cavity.
2. Feeding Difficulties
- Milk regurgitation through the nose.
- Poor suction and prolonged feeding time.
- Failure to gain weight (Due to poor feeding efficiency).
3. Speech & Language Issues
- Delayed speech development.
- Nasal speech (Hypernasality) due to air escaping through the cleft.
4. Recurrent Ear Infections (Otitis Media)
- Improper drainage of the middle ear (Due to Eustachian tube dysfunction).
- Hearing loss if chronic infections occur.
5. Dental & Orthodontic Problems
- Misaligned teeth, missing teeth, or overcrowded teeth.
- Higher risk of tooth decay due to poor oral hygiene.
Diagnosis:
1. Prenatal Diagnosis:
- Ultrasound (18-22 weeks gestation) – May detect cleft palate, though less obvious than cleft lip.
2. Postnatal Diagnosis:
- Physical Examination – Inspect the oral cavity for palatal defects.
- Feeding Assessment – Evaluate sucking and swallowing ability.
- Hearing Evaluation – Assess for middle ear dysfunction.
- Speech & Language Assessment – If the child is older.
Medical Management (Pre-Surgical Care):
1. Feeding Support:
- Use of specialized bottles (Haberman Feeder, Pigeon Bottle, Mead-Johnson Cleft Palate Nurser).
- Encourage upright feeding position to prevent milk regurgitation.
- Frequent burping to prevent aspiration.
2. Speech Therapy:
- Helps with articulation and nasal speech issues after surgical repair.
3. Hearing & Ear Care:
- Monitor for recurrent ear infections.
- Tympanostomy tube placement (If recurrent otitis media occurs).
4. Dental & Orthodontic Care:
- Early dental evaluation and orthodontic treatment to correct misaligned teeth.
Surgical Management (Definitive Treatment):
1. Timing of Surgery:
- Palatoplasty (Cleft Palate Repair) → Done at 9-18 months of age.
- Pharyngoplasty (If speech problems persist after repair) → Done at 4-6 years old.
2. Surgical Techniques:
- Von Langenbeck Repair (Most common technique).
- Furlow Double Opposing Z-Plasty (Improves speech outcomes).
3. Goals of Surgery:
- Close the cleft to separate the oral and nasal cavities.
- Improve speech articulation and feeding ability.
- Reduce risk of ear infections.
4. Postoperative Care:
- Pain management (Acetaminophen, Ibuprofen).
- Protect the surgical site (No pacifiers, straws, or hard foods).
- Use elbow restraints to prevent the child from touching the stitches.
- Monitor for signs of infection or respiratory distress.
Nursing Management:
1. Preoperative Nursing Care:
- Assess feeding difficulties and teach parents alternative feeding methods.
- Monitor for weight gain and nutritional status.
- Provide emotional support to parents (Addressing concerns about speech and appearance).
- Educate parents about the surgical procedure and postoperative care.
2. Postoperative Nursing Care:
A. Pain Management:
- Administer prescribed analgesics (Acetaminophen, Ibuprofen).
- Provide comfort measures (Rocking, Cuddling).
B. Preventing Surgical Site Complications:
- Keep the surgical site clean & dry.
- Apply antibiotic ointment as prescribed.
- Use elbow restraints to prevent the child from touching the stitches.
- Avoid pacifiers, straws, or hard foods that may disturb the sutures.
C. Feeding & Nutrition:
- Encourage spoon-feeding or cup-feeding postoperatively.
- Provide soft, pureed foods until healing is complete.
- Avoid sucking motions to prevent stress on the surgical site.
D. Monitor for Complications:
- Watch for signs of infection (Redness, Swelling, Fever).
- Monitor for breathing difficulties or airway obstruction (Rare but possible due to swelling).
E. Parental Education:
- Explain proper wound care and feeding techniques.
- Emphasize follow-up visits with the surgeon, speech therapist, and orthodontist.
- Encourage emotional bonding and psychosocial support.
Complications of Cleft Palate (If Untreated):
- Severe feeding difficulties → Malnutrition & growth retardation.
- Speech impairment → Hypernasality, delayed language development.
- Frequent ear infections → Hearing loss & learning difficulties.
- Dental abnormalities → Malocclusion, increased risk of cavities.
- Psychosocial issues → Poor self-esteem, social isolation.
Prognosis:
- With early surgical repair, feeding therapy, and speech therapy, children with cleft palate can lead a normal life.
- Most children have minimal speech impairment if treated early.
Key Points:
✔ Cleft palate is a congenital defect where the roof of the mouth fails to close properly.
✔ It can be incomplete, complete, unilateral, or bilateral.
✔ Diagnosis is done via prenatal ultrasound or postnatal physical examination.
✔ Definitive treatment is surgical repair (Palatoplasty at 9-18 months).
✔ Postoperative care includes feeding modifications, pain management, and infection prevention.
✔ Nursing care focuses on preoperative feeding support, postoperative wound care, and family education
Congenital Hypertrophic Pyloric Stenosis (HPS)
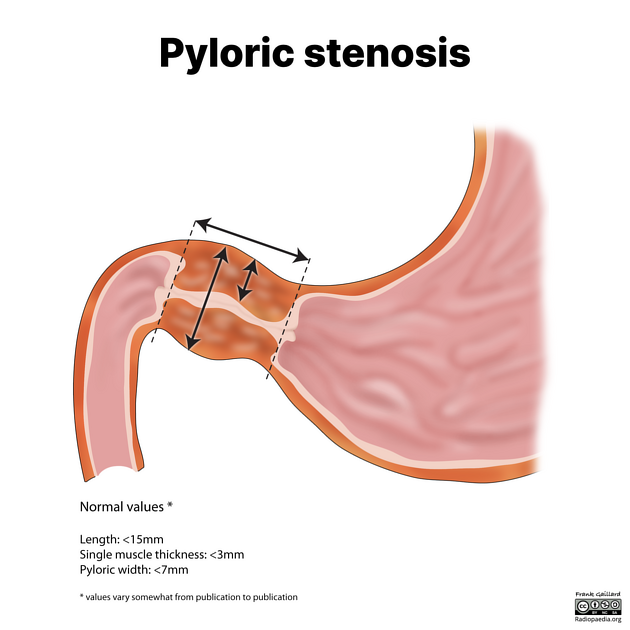
Definition:
Congenital Hypertrophic Pyloric Stenosis (HPS) is a thickening of the pyloric muscle (the muscle between the stomach and the small intestine), leading to gastric outlet obstruction. This results in progressive vomiting, dehydration, and failure to thrive in infants.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Family history (More common if parents or siblings had HPS).
- Higher incidence in first-born male infants.
- More common in Caucasian populations.
2. Environmental Factors:
- Maternal smoking during pregnancy.
- Bottle-feeding (Higher incidence than in breastfed infants).
- Use of macrolide antibiotics (Erythromycin, Azithromycin) in neonates.
Types of Hypertrophic Pyloric Stenosis:
| Type | Description |
|---|---|
| Classic HPS | Gradual hypertrophy of the pyloric muscle, leading to complete obstruction. |
| Atypical HPS | Partial obstruction with intermittent symptoms. |
Pathophysiology:
- Hypertrophy (Thickening) and Hyperplasia (Increase in cell number) of the pyloric muscle cause narrowing of the pyloric canal.
- Delayed gastric emptying → Stomach contents cannot pass into the small intestine.
- Persistent vomiting leads to:
- Dehydration
- Electrolyte imbalance (Hypochloremic metabolic alkalosis due to loss of gastric acid)
- Failure to thrive (Weight loss, Malnutrition)
Clinical Manifestations (Signs & Symptoms):
1. Classic Symptoms (Usually Appears at 3-6 Weeks of Age):
- Projectile, non-bilious vomiting after feeding.
- Hungry infant (Wants to feed again immediately after vomiting).
- Progressive weight loss, dehydration, lethargy.
2. Physical Examination Findings:
- Palpable “olive-shaped” mass in the right upper quadrant or epigastric region.
- Visible gastric peristalsis (Wave-like contractions moving from left to right).
- Sunken fontanelle, dry mucous membranes (Signs of dehydration).
Diagnosis:
1. Clinical Examination:
- Olive-like mass in the epigastrium.
- Projectile vomiting without bile (Non-bilious emesis).
2. Laboratory Findings:
- Metabolic alkalosis (↑ pH, ↑ HCO₃, ↓ Chloride).
- Hypokalemia (Low potassium levels due to vomiting).
- Elevated Blood Urea Nitrogen (BUN) (Sign of dehydration).
3. Imaging Studies:
- Ultrasound (Gold Standard):
- Thickened pyloric muscle (>3-4 mm).
- Elongated pyloric canal (>16 mm).
- Barium Swallow (If ultrasound is inconclusive):
- “String sign” – Thin streak of contrast passing through the narrowed pylorus.
Medical Management (Pre-Surgical Care):
1. Correction of Dehydration & Electrolyte Imbalance:
- IV Fluids (Normal Saline + Dextrose).
- Electrolyte Replacement (Potassium if needed).
- NPO (Nothing by mouth) until surgery is performed.
2. Gastric Decompression:
- Nasogastric (NG) tube insertion for severe vomiting.
Surgical Management (Definitive Treatment):
1. Pyloromyotomy (Ramstedt’s Procedure) – Treatment of Choice
- Procedure:
- Longitudinal incision of the hypertrophied pyloric muscle (Without cutting the mucosa).
- Allows normal passage of stomach contents into the intestine.
- Timing:
- Surgery is performed once fluid & electrolyte balance is restored.
2. Postoperative Care:
- Oral feeding resumes within 6-12 hours after surgery.
- Gradual transition from clear liquids to formula/breast milk.
- Monitor for vomiting (Common in the first 24-48 hours but should decrease).
Nursing Management:
1. Preoperative Nursing Care:
- Monitor for signs of dehydration (Dry mucous membranes, Sunken fontanelle, Poor skin turgor).
- Assess vital signs (Tachycardia, Hypotension = Severe dehydration).
- Monitor intake & output (Strict I&O charting).
- Administer IV fluids & electrolyte replacements as prescribed.
- Educate parents about the condition, treatment, and expected outcomes.
2. Postoperative Nursing Care:
- Monitor for pain (Administer acetaminophen if needed).
- Assess for surgical site infection (Redness, Swelling, Fever).
- Monitor for vomiting (Mild vomiting may persist but should gradually resolve).
- Encourage early oral feedings with small, frequent meals.
- Burp the baby frequently to prevent gas buildup.
3. Parental Education:
- Reassure parents that surgery is highly successful.
- Teach signs of dehydration and when to seek medical attention.
- Advise on proper feeding techniques post-surgery.
- Schedule follow-up visits to monitor weight gain and recovery.
Complications (If Untreated or Delayed Surgery):
- Severe dehydration & electrolyte imbalance → Shock.
- Malnutrition & failure to thrive.
- Aspiration pneumonia (If vomiting continues unchecked).
- Delayed gastric emptying (Rare, post-surgical complication).
Prognosis:
- Excellent outcome with surgery (Mortality rate <1%).
- Full recovery within a few weeks.
- Low risk of recurrence.
Key Points:
✔ Hypertrophic Pyloric Stenosis is a congenital condition causing gastric outlet obstruction.
✔ Projectile, non-bilious vomiting in a 3-6 week-old infant is the hallmark symptom.
✔ Diagnosis is confirmed by ultrasound (Thickened pyloric muscle).
✔ IV fluid resuscitation is crucial before surgery (Pyloromyotomy).
✔ Postoperative care focuses on feeding reintroduction and monitoring for complications.
Hirschsprung’s Disease (Congenital Megacolon)
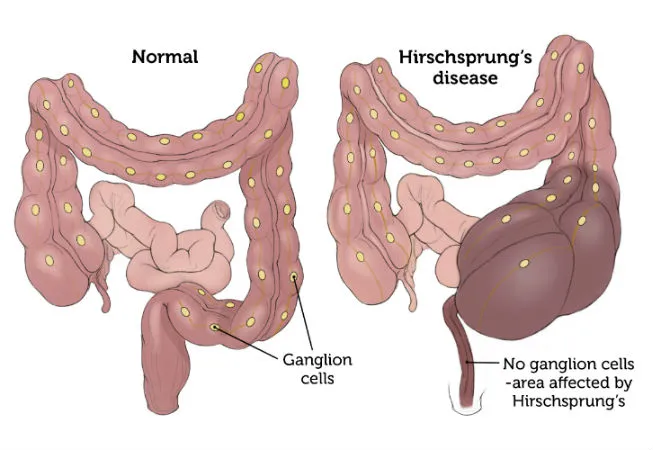
Definition:
Hirschsprung’s Disease (HD) is a congenital disorder characterized by the absence of ganglion cells in the myenteric (Auerbach) and submucosal (Meissner) plexuses of the colon, leading to failure of peristalsis and intestinal obstruction. It results in megacolon (distended colon) due to functional obstruction.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Autosomal dominant or recessive inheritance.
- Mutations in the RET proto-oncogene.
- Associated with syndromes (Down syndrome, Waardenburg syndrome).
2. Environmental Factors:
- Fetal hypoxia affecting enteric neural crest migration.
- Maternal exposure to drugs or toxins.
Types of Hirschsprung’s Disease:
| Type | Description |
|---|---|
| Short-Segment Hirschsprung’s Disease | Affects the rectosigmoid region (Most common). |
| Long-Segment Hirschsprung’s Disease | Extends beyond the sigmoid colon. |
| Total Colonic Aganglionosis | Entire colon is affected (Rare). |
| Ultra-Short Segment Hirschsprung’s Disease | Only affects the anus or lower rectum. |
Pathophysiology:
- Failure of neural crest cells to migrate during fetal development → Leads to absence of ganglion cells in the colon.
- Lack of ganglion cells leads to failure of peristalsis → Functional obstruction in the affected segment.
- Proximal colon dilates (Megacolon) due to stool accumulation, while the distal part remains narrow and contracted.
- Intestinal obstruction leads to abdominal distension, failure to pass meconium, and chronic constipation.
Clinical Manifestations (Signs & Symptoms):
1. Neonatal Period (Severe Cases)
- Failure to pass meconium within 24-48 hours of birth.
- Abdominal distension.
- Bilious vomiting (Greenish vomit due to intestinal obstruction).
- Poor feeding, irritability.
2. Infancy & Childhood (Milder Cases)
- Chronic constipation (Since birth).
- Ribbon-like, foul-smelling stools.
- Failure to thrive (Poor weight gain).
- Recurrent episodes of enterocolitis (Diarrhea, Fever, Abdominal distension).
3. Severe Cases (Complications)
- Hirschsprung-Associated Enterocolitis (HAEC) → Life-threatening complication with fever, explosive diarrhea, and sepsis.
- Toxic Megacolon → Extreme dilation of the colon with risk of perforation.
Diagnosis:
1. Clinical Examination:
- Rectal Exam: Tight anal sphincter with no stool in the rectum.
2. Laboratory Tests:
- Complete Blood Count (CBC) – May show signs of infection (In enterocolitis cases).
- Serum Electrolytes – May show dehydration due to vomiting or diarrhea.
3. Imaging Studies:
- Abdominal X-ray (Plain Film):
- Dilated colon (Megacolon) proximal to the affected segment.
- Absence of air in the rectum.
- Barium Enema:
- “Transition zone” between the dilated proximal colon and narrow distal colon.
4. Definitive Diagnosis:
- Rectal Biopsy (Gold Standard):
- Absence of ganglion cells in the affected segment.
- Increased acetylcholinesterase activity (Confirms Hirschsprung’s disease).
- Anorectal Manometry:
- Failure of the internal anal sphincter to relax when the rectum is distended.
Medical Management (Pre-Surgical Care):
- Bowel decompression using rectal irrigation.
- IV fluids & electrolyte correction.
- Nasogastric tube insertion (For gastric decompression).
- Antibiotics if enterocolitis is present.
Surgical Management (Definitive Treatment):
1. Pull-Through Procedures (Definitive Treatment)
- Swenson Procedure: Removes the affected segment and brings the healthy colon down.
- Duhamel Procedure: Bypasses the aganglionic segment using a side-to-side anastomosis.
- Soave Procedure: Leaves a part of the muscular layer intact, removing the inner aganglionic layer.
2. Staged Surgery (For Severe Cases)
- Colostomy (Temporary diversion of stool)
- Performed first to allow bowel healing.
- Definitive Pull-Through Surgery (After 6-12 months).
- Colostomy Closure (Final stage).
Nursing Management:
1. Preoperative Nursing Care:
- Monitor for signs of enterocolitis (Diarrhea, Fever, Abdominal distension).
- Ensure proper hydration (IV fluids if needed).
- Administer antibiotics if infection is present.
- Teach parents about colostomy care (If a staged surgery is planned).
2. Postoperative Nursing Care:
A. Pain Management
- Administer analgesics as prescribed (Acetaminophen, Ibuprofen).
- Provide comfort measures (Swaddling, Holding).
B. Monitoring for Complications
- Monitor for signs of infection (Fever, Redness, Discharge at surgical site).
- Watch for bowel obstruction signs (Absent bowel sounds, Distension, Vomiting).
C. Colostomy Care (If Performed)
- Educate parents on colostomy bag changes.
- Assess for skin irritation around the colostomy site.
- Ensure proper stool output (Avoid dehydration).
D. Feeding & Nutrition
- Introduce oral feeds gradually after surgery.
- Encourage high-fiber diet (For older children to prevent constipation).
Complications of Hirschsprung’s Disease:
- Hirschsprung-Associated Enterocolitis (HAEC)
- Severe diarrhea, Fever, Sepsis, Toxic Megacolon (Medical Emergency).
- Bowel Obstruction & Perforation
- Requires urgent surgical intervention.
- Short Bowel Syndrome (If a large segment is removed).
- Constipation & Fecal Incontinence (Long-term issues after surgery).
Prognosis:
- Excellent prognosis if treated early with surgery.
- Most children recover completely, but some may have mild constipation.
- Severe cases with complications (Toxic megacolon, Perforation) may require prolonged treatment.
Key Points:
✔ Hirschsprung’s Disease is a congenital absence of ganglion cells in the colon, leading to functional obstruction.
✔ Signs include failure to pass meconium, chronic constipation, and abdominal distension.
✔ Definitive diagnosis is by rectal biopsy (Absence of ganglion cells).
✔ Surgical treatment (Pull-through procedure) is the cure.
✔ Nursing care focuses on preoperative hydration, postoperative monitoring, and colostomy care if needed.
Anorectal Malformations (Congenital Anorectal Anomalies)
Definition:
Anorectal malformations (ARMs) are congenital defects where the anus and rectum fail to develop properly, leading to abnormal positioning or absence of the anal opening. These anomalies may occur alone or as part of syndromic conditions and can involve rectal atresia, stenosis, fistulas, or an imperforate anus.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Sporadic mutations in genes responsible for embryonic development.
- Familial inheritance in some cases.
- Associated syndromes (VACTERL association, Down syndrome, Currarino syndrome).
2. Environmental Factors:
- Maternal drug exposure (Thalidomide, Retinoic acid).
- Folic acid deficiency during pregnancy.
- Maternal infections affecting fetal development.
Types of Anorectal Malformations:
1. Based on Severity & Location:
| Type | Description |
|---|---|
| Imperforate Anus | Absence of a normal anal opening. |
| Anal Stenosis | Narrowing of the anal opening, causing difficulty in passing stool. |
| Rectal Atresia | Complete absence of a rectal opening, requiring surgical correction. |
| Rectoperineal Fistula | Abnormal connection between the rectum and perineum. |
| Rectovestibular Fistula (Females) | Rectum opens into the vestibule of the vagina. |
| Rectourethral Fistula (Males) | Rectum is connected to the urethra. |
| Cloacal Malformation (Females) | Single opening for the urinary, genital, and rectal tracts. |
2. Based on High vs. Low Anomalies:
| Type | Description | Common Associations |
|---|---|---|
| Low Anomalies | Anus is close to the perineum, with minor fistulas. | Good prognosis, easier surgical repair. |
| High Anomalies | Rectum is high in the pelvis, requiring extensive reconstruction. | Associated with VACTERL anomalies (Vertebral, Anorectal, Cardiac, Tracheoesophageal, Renal, Limb defects). |
Pathophysiology:
- During the 4th-7th week of fetal development, the urorectal septum fails to separate the rectum from the urinary and genital structures.
- This leads to:
- Incomplete formation of the anus and rectum.
- Abnormal connections (fistulas) between the rectum and nearby organs.
- Functional consequences:
- Intestinal obstruction due to lack of a normal anal opening.
- Fecal incontinence if sphincter muscles are affected.
- Urinary or genital complications if fistulas are present.
Clinical Manifestations (Signs & Symptoms):
1. Neonatal Symptoms (First 24-48 Hours)
- Failure to pass meconium within 24-48 hours of birth.
- Abdominal distension (Due to obstruction).
- Vomiting (May be bilious if associated with intestinal obstruction).
- Absence of a visible anal opening.
2. Symptoms in Mild Cases
- Narrowed anal opening with difficulty in passing stools (Anal stenosis).
- Passing stool through abnormal openings (Fistula to perineum, vagina, or urethra).
- Frequent urinary tract infections (Due to recto-urinary fistulas).
3. Associated Anomalies (If Part of a Syndrome)
- Renal abnormalities (Hydronephrosis, Vesicoureteral reflux).
- Cardiac defects (VSD, ASD, Tetralogy of Fallot).
- Spinal anomalies (Sacral agenesis, Spina bifida).
Diagnosis:
1. Physical Examination:
- Inspect perineal region for absence of an anal opening or presence of fistulas.
- Assess abdominal distension and failure to pass meconium.
2. Imaging Studies:
- Abdominal X-ray (Cross-table lateral view)
- Determines distance of the rectum from the perineum (Differentiates high vs. low anomalies).
- Ultrasound
- Assesses associated renal and bladder anomalies.
- Contrast Studies (Fistulogram, Voiding Cystourethrogram – VCUG)
- Detects rectal fistulas (If stool passes through the urethra or vagina).
- MRI/CT Scan
- Evaluates spinal anomalies (If part of VACTERL syndrome).
Medical Management (Pre-Surgical Care):
- NPO (Nothing by mouth) to prevent bowel distension.
- IV fluids & Electrolyte correction.
- Nasogastric (NG) tube decompression (If severe abdominal distension).
- Antibiotics (If risk of urinary tract infections due to fistula).
- Parental education regarding the surgical plan.
Surgical Management (Definitive Treatment):
1. Low Anomalies (Mild Cases)
- Anoplasty → Simple surgical correction of anal stenosis.
- Dilation Therapy → Used postoperatively to prevent narrowing.
2. High Anomalies (Severe Cases)
- Staged Repair Approach:
- Colostomy (First stage, performed at birth) → Diverts stool to allow intestinal decompression.
- Definitive Pull-Through Surgery (2-6 months later) → Creates a new anus and reconnects the rectum.
- Colostomy Closure (Final stage, after healing).
- Posterior Sagittal Anorectoplasty (PSARP, “Pull-Through” Surgery)
- Most commonly used to reconstruct the anal canal.
3. Complex Anomalies (Cloacal Malformation, Fistulas)
- Multistage surgery required, involving urologists & pediatric surgeons.
- Urethral and genital reconstruction if needed.
Nursing Management:
1. Preoperative Nursing Care:
- Assess for signs of bowel obstruction (Vomiting, Distension, No meconium).
- Monitor for urinary symptoms (If recto-urinary fistula is present).
- Administer IV fluids to maintain hydration.
- Insert NG tube for gastric decompression.
- Prepare parents for possible colostomy and future surgical corrections.
2. Postoperative Nursing Care:
A. Pain Management
- Administer analgesics as prescribed (Acetaminophen, Ibuprofen).
- Use non-pharmacological comfort measures (Swaddling, Holding).
B. Monitoring for Complications
- Assess surgical site for signs of infection (Redness, Swelling, Discharge).
- Monitor for bowel movements (Ensure normal stool passage post-surgery).
- Watch for signs of bowel obstruction (Vomiting, No stool output).
C. Colostomy Care (If Performed)
- Teach parents proper colostomy bag changes.
- Monitor for skin irritation around the colostomy site.
- Ensure proper stool output (Avoid dehydration).
D. Feeding & Nutrition
- Start oral feedings gradually postoperatively.
- Encourage fiber-rich diet (To prevent constipation post-repair).
3. Parental Education:
- Teach signs of constipation and obstruction.
- Emphasize the need for regular follow-ups with pediatric surgery.
- Discuss future continence management (Some children may need long-term bowel training).
Complications of Anorectal Malformations:
- Bowel obstruction (If untreated).
- Chronic constipation & incontinence (If sphincter function is affected).
- Recurrent urinary tract infections (If recto-urinary fistulas remain untreated).
- Colostomy-related complications (Skin irritation, Leakage).
Prognosis:
- Low anomalies have an excellent prognosis with early surgical correction.
- High anomalies may require long-term management for continence.
- Multidisciplinary care is needed for complex cases (Urologists, Surgeons, Gastroenterologists).
Key Points:
✔ Anorectal malformations are congenital defects in the anus and rectum, leading to functional obstruction.
✔ Failure to pass meconium, abdominal distension, and abnormal anal opening are key signs.
✔ Diagnosis is confirmed with imaging and rectal biopsy if needed.
✔ Surgical correction (Anoplasty, Pull-through, or Staged repair) is the definitive treatment.
✔ Nursing care focuses on preoperative bowel management, postoperative monitoring, and parental education.
Anorectal Malformation (Congenital Anorectal Anomalies)
Definition:
Anorectal malformations (ARMs) are congenital birth defects in which the anus and rectum do not develop properly. This results in partial or complete obstruction of the normal passage of stool due to absence, abnormal positioning, or connection of the rectum to the urinary or genital structures (fistulas).
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Sporadic mutations affecting embryonic development.
- Familial inheritance (Occurs more frequently in families with a history of ARM).
- Syndromic associations (VACTERL association, Currarino syndrome, Down syndrome).
2. Environmental Factors:
- Maternal exposure to teratogens (Thalidomide, Retinoic acid).
- Folic acid deficiency during pregnancy.
- Maternal infections or diabetes.
Types of Anorectal Malformations:
1. Based on Anatomic Classification:
| Type | Description |
|---|---|
| Imperforate Anus | Absence of an anal opening. |
| Anal Stenosis | Narrowed anal opening, causing difficulty in passing stool. |
| Rectal Atresia | Complete blockage of the rectum with no connection to the anus. |
| Rectoperineal Fistula | The rectum opens into the perineum instead of the anus. |
| Rectovestibular Fistula (Females) | The rectum opens into the vaginal vestibule. |
| Rectourethral Fistula (Males) | The rectum connects to the urethra, causing stool to pass through urine. |
| Cloacal Malformation (Females) | The rectum, urinary tract, and genital tract form a single opening. |
2. Based on Severity & Location:
| Type | Description | Common Associations |
|---|---|---|
| Low Anomalies | Anus is near the normal location, with minor fistulas. | Good prognosis, easier surgical repair. |
| High Anomalies | Rectum is higher in the pelvis, requiring major surgery. | Associated with VACTERL anomalies (Vertebral, Anorectal, Cardiac, Tracheoesophageal, Renal, Limb defects). |
Pathophysiology:
- During the 4th-7th week of fetal development, the urorectal septum fails to separate the rectum from the urinary and genital structures.
- This results in:
- Absence of an anal opening or abnormal positioning of the rectum.
- Fistulas connecting the rectum to the urinary or genital tracts.
- Functional consequences:
- Intestinal obstruction → Due to incomplete or absent anal opening.
- Urinary tract infections → If the rectum is connected to the urethra or bladder.
- Fecal incontinence → If the anal sphincters are underdeveloped.
Clinical Manifestations (Signs & Symptoms):
1. Neonatal Symptoms (Within 24-48 Hours)
- Failure to pass meconium (First stool) within 24-48 hours.
- Abdominal distension due to intestinal obstruction.
- Bilious or non-bilious vomiting.
- Absence or abnormal positioning of the anal opening.
- Passage of stool through the urethra or vagina (Indicates a fistula).
- Frequent urinary tract infections (If recto-urinary fistula is present).
2. Symptoms in Mild or Undiagnosed Cases
- Chronic constipation.
- Difficulty passing stool (Anal stenosis).
- Fecal soiling or incontinence in older children.
- Perianal skin irritation due to abnormal stool passage.
3. Associated Anomalies (VACTERL Syndrome)
- Vertebral defects (Spinal abnormalities, scoliosis).
- Cardiac anomalies (VSD, ASD, Tetralogy of Fallot).
- Tracheoesophageal fistula (TEF, abnormal connection between the esophagus and trachea).
- Renal malformations (Hydronephrosis, vesicoureteral reflux).
- Limb anomalies (Radial dysplasia, missing fingers/toes).
Diagnosis:
1. Physical Examination:
- Inspection of the perineal region for absence of an anal opening or fistula.
- Digital rectal examination (In low-type anomalies, may feel a narrowed anal canal).
2. Imaging Studies:
- Abdominal X-ray (Cross-table lateral view)
- Determines the distance of the rectum from the perineum (Differentiates high vs. low anomalies).
- Ultrasound (Evaluates renal and bladder anomalies).
- Contrast Studies (Fistulogram, Voiding Cystourethrogram – VCUG)
- Detects rectal fistulas (If stool passes through the urethra or vagina).
- MRI/CT Scan (To assess pelvic muscles and spinal cord defects).
3. Definitive Diagnosis:
- Rectal Biopsy (If Hirschsprung’s disease is suspected).
Medical Management (Pre-Surgical Care):
- NPO (Nothing by mouth) to prevent bowel distension.
- IV fluids & electrolyte correction.
- Nasogastric (NG) tube insertion for gastric decompression.
- Antibiotics if urinary tract infection is present.
- Parental education on the surgical plan.
Surgical Management (Definitive Treatment):
1. Low Anomalies (Mild Cases)
- Anoplasty (Surgical creation of a normal anus).
- Dilation Therapy (Postoperative care to prevent narrowing).
2. High Anomalies (Severe Cases)
- Staged Repair Approach:
- Colostomy (Temporary diversion of stool) – Performed at birth to allow bowel healing.
- Definitive Pull-Through Surgery (2-6 months later) – Creates a new anus and reconnects the rectum.
- Colostomy Closure (Final stage, after healing).
- Posterior Sagittal Anorectoplasty (PSARP, “Pull-Through” Surgery)
- The most common method for reconstructing the anal canal.
3. Complex Anomalies (Cloacal Malformation, Fistulas)
- Multistage surgery required involving pediatric surgeons and urologists.
- Urogenital reconstruction may be needed if abnormalities extend to the reproductive system.
Nursing Management:
1. Preoperative Nursing Care:
- Monitor for signs of bowel obstruction (Vomiting, Distension, No meconium).
- Assess urinary symptoms (If recto-urinary fistula is present).
- Administer IV fluids to maintain hydration.
- Insert NG tube for gastric decompression.
- Prepare parents for possible colostomy and future surgeries.
2. Postoperative Nursing Care:
A. Pain Management
- Administer analgesics (Acetaminophen, Ibuprofen).
- Use non-pharmacological comfort measures (Swaddling, Holding).
B. Monitoring for Complications
- Assess surgical site for signs of infection (Redness, Swelling, Discharge).
- Monitor for bowel movements (Ensure normal stool passage post-surgery).
- Watch for signs of bowel obstruction (Absent bowel sounds, Distension, Vomiting).
C. Colostomy Care (If Performed)
- Teach parents proper colostomy care.
- Monitor for skin irritation around the colostomy site.
- Ensure proper stool output (Avoid dehydration).
D. Feeding & Nutrition
- Start oral feeds gradually postoperatively.
- Encourage a fiber-rich diet (For older children to prevent constipation).
3. Parental Education:
- Teach signs of constipation and obstruction.
- Emphasize the need for regular follow-ups with pediatric surgery.
- Discuss long-term bowel management for continence.
Complications of Anorectal Malformations:
- Bowel obstruction (If untreated).
- Chronic constipation & incontinence (If sphincter function is affected).
- Recurrent urinary tract infections (If recto-urinary fistulas remain untreated).
- Colostomy-related complications (Skin irritation, Leakage).
Prognosis:
- Low anomalies have an excellent prognosis with early surgical correction.
- High anomalies may require long-term bowel management.
- Multidisciplinary care (Surgeons, urologists, gastroenterologists) is essential.
Abdominal Wall Defects in Newborns
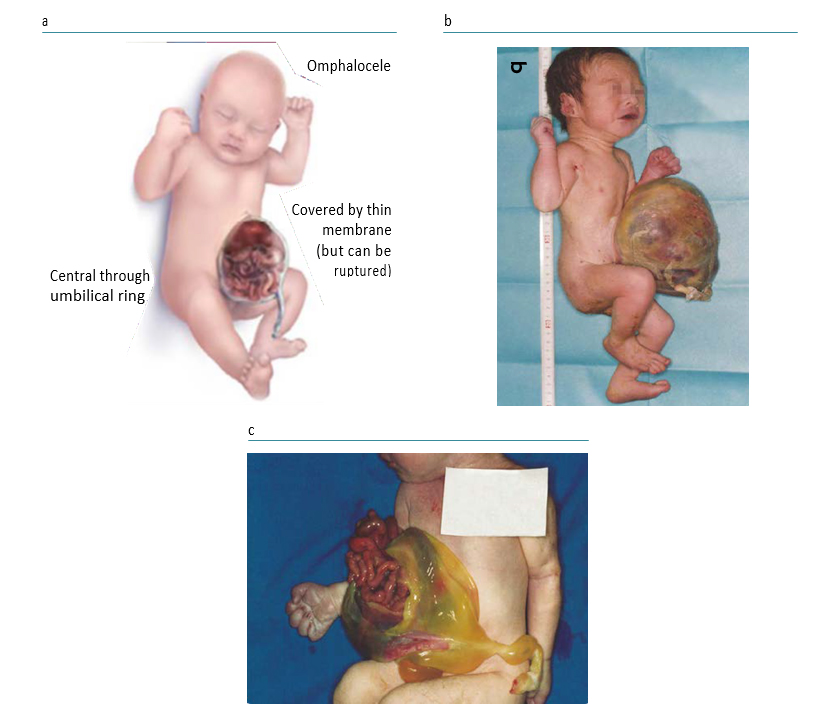
Definition:
Abdominal wall defects are congenital anomalies where the fetal abdominal wall fails to close properly during development, causing abdominal organs (intestines, liver, stomach) to protrude outside the body. The two main types of abdominal wall defects are gastroschisis and omphalocele.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Chromosomal abnormalities (Trisomy 13, 18, and Beckwith-Wiedemann Syndrome – Associated with Omphalocele).
- Family history of congenital abdominal defects.
2. Environmental Factors:
- Maternal smoking, alcohol, or illicit drug use (Cocaine, Methamphetamines).
- Exposure to teratogens (Pesticides, Radiation).
- Maternal nutritional deficiencies (Folic acid, Zinc deficiency).
Types of Abdominal Wall Defects:
| Type | Description | Common Associations |
|---|---|---|
| Gastroschisis | Intestines protrude through a defect to the right of the umbilicus, without a protective sac. | No chromosomal abnormalities, associated with maternal smoking and young maternal age. |
| Omphalocele | Intestines, liver, or stomach protrude through the umbilical ring, covered by a thin membrane sac. | Associated with chromosomal abnormalities (Trisomy 13, 18, Beckwith-Wiedemann Syndrome, congenital heart defects). |
Pathophysiology:
- During fetal development (4th-8th week), the abdominal organs fail to return to the abdominal cavity from the umbilical cord.
- Gastroschisis occurs due to vascular disruption, leading to a hole beside the umbilicus, with exposed intestines.
- Omphalocele occurs due to failure of lateral body folds to fuse, leaving the intestines covered by a thin membrane.
- Consequences of Abdominal Organ Exposure:
- Risk of infection (If sac ruptures in Omphalocele or due to exposed organs in Gastroschisis).
- Heat loss and dehydration (Due to evaporative fluid loss from exposed intestines).
- Malrotation of the intestines (Leads to bowel obstruction and ischemia).
Clinical Manifestations (Signs & Symptoms):
1. Gastroschisis:
- Intestines protrude through a defect to the right of the umbilicus.
- No protective sac covering the exposed intestines.
- Bowel appears thickened, inflamed, and edematous.
- Risk of bowel damage due to exposure to amniotic fluid.
2. Omphalocele:
- Intestines, stomach, or liver protrude through the umbilical cord.
- Organs are enclosed in a thin, transparent sac.
- High risk of associated congenital defects (Cardiac anomalies, Chromosomal syndromes).
- Rupture of the sac increases the risk of infection and bowel damage.
3. Complications of Both Conditions:
- Bowel obstruction (Due to twisted or malrotated intestines).
- Intestinal perforation and necrosis (If blood supply is compromised).
- Respiratory distress (Due to abdominal pressure affecting lung expansion).
Diagnosis:
1. Prenatal Diagnosis:
- Ultrasound (At 12-14 weeks gestation)
- Detects herniated bowel loops outside the fetal abdomen.
- Elevated Alpha-Fetoprotein (AFP) Levels in Maternal Blood
- Suggests abdominal wall defects, neural tube defects, or other anomalies.
2. Postnatal Diagnosis:
- Physical Examination
- Identifies location of the defect, presence of a protective sac, and severity of organ involvement.
- X-ray or Abdominal Ultrasound
- Assesses intestinal malrotation, obstruction, or additional anomalies.
- Echocardiogram and Genetic Testing
- Performed for omphalocele due to high association with heart defects and chromosomal abnormalities.
Medical Management (Pre-Surgical Care):
1. Immediate Stabilization at Birth:
- Keep the infant in a sterile, warm environment (Prevent hypothermia).
- Cover exposed intestines with a sterile, moist dressing (Prevent infection and fluid loss).
- Place the lower half of the baby in a plastic bowel bag or sterile wrap (Reduces heat and fluid loss).
- Position the baby supine (Prevent kinking of the bowel).
2. Fluid & Electrolyte Management:
- IV Fluids & Electrolytes to maintain hydration.
- Monitor urine output & serum electrolytes (Prevent dehydration).
3. Preventing Infection:
- Broad-spectrum antibiotics (Prevent peritonitis and sepsis).
- Monitor for signs of infection (Fever, Tachycardia, Wound redness).
4. Respiratory & Cardiac Support:
- Administer Oxygen (If respiratory distress is present).
- Nasogastric Tube (For gastric decompression & reducing bowel pressure).
Surgical Management (Definitive Treatment):
1. Gastroschisis Repair:
- Primary Repair (If small defect)
- Immediate closure of the abdominal wall after reducing the intestines into the abdomen.
- Staged Repair (If large defect or compromised bowel)
- Silo Placement:
- A plastic silo bag is used to enclose the intestines and gradually return them into the abdomen over several days.
- Final closure is done once the abdominal cavity has expanded.
- Silo Placement:
2. Omphalocele Repair:
- Primary Closure (If small and no other defects present).
- Staged Closure (If large defect or lung immaturity).
- Non-operative Management (For Giant Omphalocele) → Uses topical agents (Silver Sulfadiazine) to promote epithelialization, delaying surgery.
Postoperative Considerations:
- Pain management (Acetaminophen, Opioids if severe).
- Slow reintroduction of oral feeds (IV Nutrition until bowel function returns).
- Monitor for abdominal compartment syndrome (Due to increased intra-abdominal pressure post-repair).
Nursing Management:
1. Preoperative Nursing Care:
- Monitor for respiratory distress (Due to abdominal pressure).
- Assess hydration and urine output (Prevent hypovolemia).
- Sterile dressing care (To keep bowel moist and protected).
- Administer IV fluids & antibiotics.
2. Postoperative Nursing Care:
A. Pain Management
- Administer prescribed analgesics (Acetaminophen, Morphine if needed).
- Provide comfort measures (Swaddling, Holding).
B. Monitoring for Complications
- Monitor for signs of infection (Fever, Increased WBC count, Redness at incision site).
- Assess for bowel function return (Monitor for stool passage, Bowel sounds).
- Watch for signs of bowel obstruction (Vomiting, Distension, Absent bowel sounds).
C. Nutrition & Feeding Support
- NPO (Nothing by mouth) until bowel function returns.
- Total Parenteral Nutrition (TPN) through IV until enteral feeding is tolerated.
- Gradual introduction of oral feeds (Start with breast milk or formula).
D. Parental Education & Support
Complications of Abdominal Wall Defects:
- Bowel Obstruction & Adhesions (Post-Surgical).
- Sepsis (If intestines are exposed for long periods).
- Short Bowel Syndrome (If large portions of bowel are removed).
- Respiratory Failure (If the repaired abdomen compresses the lungs).
- Abdominal Compartment Syndrome (Due to increased intra-abdominal pressure).
Prognosis:
- Gastroschisis: Good prognosis if repaired early, with 90-95% survival rate.
- Omphalocele: Prognosis depends on associated genetic conditions, but isolated cases have a 80-90% survival rate.
Congenital Hernia in Children
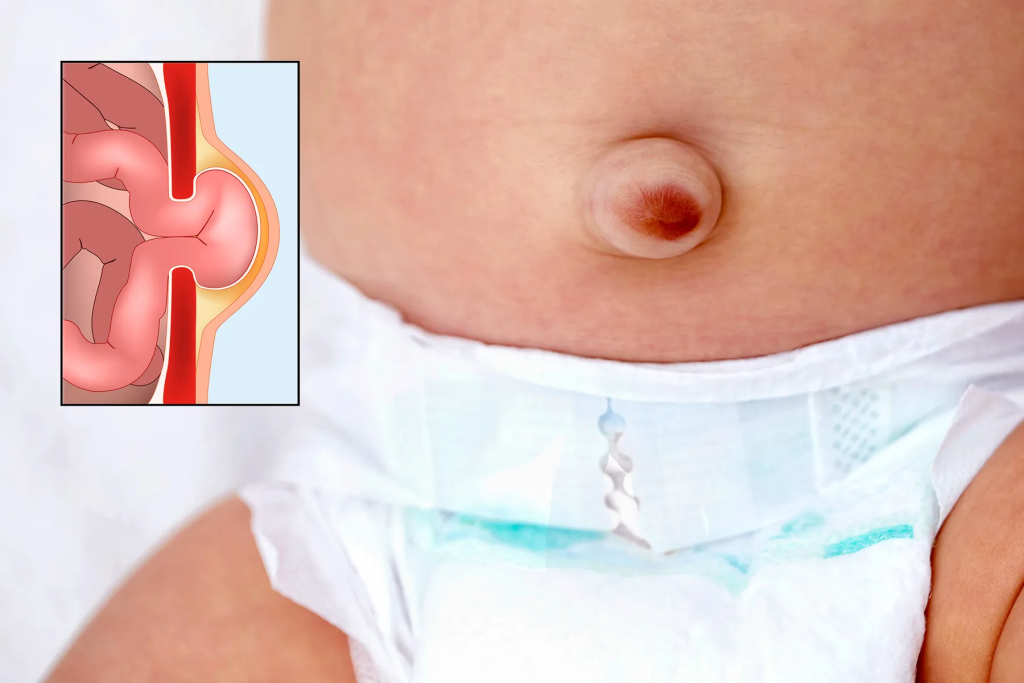
Definition:
A congenital hernia is a condition present at birth where an internal organ, such as the intestine, protrudes through a weak spot or defect in the abdominal wall or diaphragm. This can cause intestinal obstruction, respiratory issues, or organ strangulation if untreated.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Familial predisposition to hernias.
- Chromosomal abnormalities (Trisomy 21, 18, 13, Turner Syndrome).
2. Developmental Factors:
- Incomplete closure of the diaphragm or abdominal wall during fetal development.
- Failure of the umbilical ring to close completely (Umbilical hernia).
3. Environmental Factors:
- Maternal smoking, alcohol, or drug use.
- Low birth weight and preterm birth (Weakened abdominal muscles).
Types of Congenital Hernias:
| Type | Description | Location |
|---|---|---|
| Diaphragmatic Hernia (Congenital Diaphragmatic Hernia – CDH) | Abdominal organs move into the chest cavity due to a defect in the diaphragm. | Thoracic cavity (Mostly left side) |
| Umbilical Hernia | Part of the intestine protrudes through the umbilical ring due to weak abdominal muscles. | Near the navel |
| Inguinal Hernia | Intestines protrude through the inguinal canal due to an open processus vaginalis. | Groin area |
| Hiatal Hernia | Part of the stomach moves up through the diaphragm into the chest. | Esophageal opening in the diaphragm |
Pathophysiology:
- Failure of fetal development leads to a defect in the abdominal wall, diaphragm, or inguinal canal.
- Abdominal contents protrude through the defect due to increased intra-abdominal pressure.
- Depending on the hernia type, complications include:
- Diaphragmatic hernia → Respiratory distress due to lung compression.
- Umbilical or inguinal hernia → Intestinal obstruction or strangulation.
- Hiatal hernia → Acid reflux and aspiration pneumonia.
Clinical Manifestations (Signs & Symptoms):
1. Congenital Diaphragmatic Hernia (CDH)
- Severe respiratory distress at birth (Cyanosis, Tachypnea, Retractions).
- Scaphoid (sunken) abdomen due to displaced abdominal contents.
- Absent or diminished breath sounds on the affected side.
- Mediastinal shift on chest X-ray.
2. Umbilical Hernia
- Soft, bulging mass near the navel (More noticeable when crying).
- Easily reducible (Can be pushed back).
- Usually asymptomatic unless strangulated.
3. Inguinal Hernia
- Swelling in the groin or scrotum (More pronounced during crying or straining).
- Pain or discomfort in older infants and children.
- Signs of strangulation (Severe pain, redness, vomiting, no reducibility).
4. Hiatal Hernia
- Frequent regurgitation or vomiting.
- Feeding difficulties, irritability.
- Aspiration pneumonia due to reflux.
Diagnosis:
1. Physical Examination:
- Inspection & Palpation – Identifies location and reducibility of the hernia.
2. Imaging Studies:
- Chest X-ray (For CDH) → Shows bowel loops in the thoracic cavity and lung underdevelopment.
- Ultrasound (For inguinal & umbilical hernias) → Confirms herniated bowel.
- Barium Swallow (For hiatal hernia) → Detects stomach displacement and reflux.
3. Laboratory Tests:
- Blood gases (For CDH) → Shows respiratory acidosis due to lung compression.
Medical Management (Pre-Surgical Care):
- Congenital diaphragmatic hernia (CDH) requires immediate stabilization.
- In cases of small umbilical or inguinal hernias, watchful waiting may be advised.
1. Immediate Stabilization for CDH:
- Endotracheal intubation & ventilation (Avoid bag-mask ventilation to prevent stomach inflation).
- NG tube decompression (Prevents further lung compression).
- IV fluids & electrolyte correction.
- Sedation & muscle relaxants (Minimizes respiratory distress).
2. Observation for Small Umbilical Hernias:
- Most close spontaneously by age 3-4 years.
- If larger than 2 cm or persists beyond 5 years, surgery is recommended.
3. Manual Reduction for Inguinal Hernia (If Not Strangulated):
- Performed by a trained healthcare provider.
- Surgical repair is still needed to prevent recurrence.
Surgical Management (Definitive Treatment):
1. Congenital Diaphragmatic Hernia Repair:
- Surgical closure of the diaphragmatic defect and returning organs to the abdomen.
- If severe lung hypoplasia, ECMO (Extracorporeal Membrane Oxygenation) may be required before surgery.
2. Umbilical Herniorrhaphy (Umbilical Hernia Repair):
- Performed if hernia is large, symptomatic, or persists beyond 4-5 years.
3. Inguinal Herniorrhaphy (Inguinal Hernia Repair):
- Immediate surgery (If strangulation risk is high).
- Laparoscopic or open repair by closing the patent processus vaginalis.
4. Hiatal Hernia Repair (Fundoplication):
- For severe cases with reflux, fundoplication (wrapping part of the stomach around the esophagus) is done.
Nursing Management:
1. Preoperative Nursing Care:
- Monitor for respiratory distress (CDH cases).
- Keep NPO (Nothing by mouth) if surgery is planned.
- Administer IV fluids & electrolytes as prescribed.
- Provide pain relief and comfort measures.
- Educate parents on surgical procedures and expected outcomes.
2. Postoperative Nursing Care:
A. Monitoring for Complications:
- Assess for respiratory distress post-CDH repair.
- Monitor for wound infection (Redness, Swelling, Drainage).
- Watch for signs of bowel obstruction (Abdominal distension, No stool passage).
B. Pain Management:
- Administer analgesics (Acetaminophen, Ibuprofen, or Opioids if needed).
- Encourage non-pharmacological comfort measures.
C. Feeding & Nutrition:
- Gradual reintroduction of oral feeds post-surgery.
- Monitor for feeding intolerance or reflux (Hiatal hernia cases).
- For CDH, infants may require supplemental oxygen and nutrition support.
D. Parental Education:
- Teach parents signs of hernia recurrence.
- Provide care instructions for wound healing and activity restrictions post-surgery.
- Instruct parents to seek immediate care if symptoms of strangulation appear (Severe pain, Vomiting, Non-reducible swelling).
Complications of Congenital Hernias:
- Respiratory Failure (In CDH cases with severe lung underdevelopment).
- Strangulated Hernia (Cut-off blood supply leading to necrosis).
- Bowel Obstruction (If intestines are trapped in a hernia sac).
- Post-Surgical Adhesions or Recurrence of Hernia.
Prognosis:
- CDH survival rate depends on lung development (60-80%).
- Umbilical hernias resolve spontaneously in most cases.
- Inguinal hernias require surgery but have excellent outcomes.
- Hiatal hernias may need lifelong reflux management.
Key Points:
✔ Congenital hernias occur due to failure of abdominal or diaphragmatic closure in fetal development.
✔ Types include CDH, Umbilical, Inguinal, and Hiatal Hernias.
✔ CDH requires immediate respiratory support and surgical correction.
✔ Small umbilical hernias often resolve spontaneously, while inguinal hernias require surgery.
✔ Postoperative care focuses on respiratory support, pain management, and feeding progression.
Gastro-intestinal system:Congenital hernia-definition,etiology,types,pathophysiology,diagnosis,clinical menifestation,medical management,surgical management,nursing management in details
Gastroenteritis in Children
Definition:
Gastroenteritis is an inflammation of the stomach and intestines, leading to diarrhea, vomiting, abdominal pain, and dehydration. It is commonly caused by infections (viral, bacterial, or parasitic) or non-infectious conditions (food intolerance, toxins, medications).
Etiology (Causes & Risk Factors):
1. Infectious Causes (Most Common)
A. Viral Gastroenteritis (Most Common Cause in Children)
- Rotavirus (Most common worldwide, severe in infants).
- Norovirus (Highly contagious, common in outbreaks).
- Adenovirus, Astrovirus (Mild gastroenteritis).
B. Bacterial Gastroenteritis
- Escherichia coli (E. coli) (Traveler’s diarrhea, Shiga-toxin-producing strains cause Hemolytic Uremic Syndrome – HUS).
- Salmonella (Foodborne, causes fever and bloody diarrhea).
- Shigella (Severe diarrhea, high fever, and dysentery).
- Campylobacter jejuni (Contaminated poultry, causes bloody diarrhea).
- Clostridium difficile (Antibiotic-associated diarrhea).
C. Parasitic Gastroenteritis
- Giardia lamblia (Contaminated water, causes prolonged diarrhea).
- Cryptosporidium (Immunocompromised children).
2. Non-Infectious Causes
- Food allergies (Milk, soy intolerance in infants).
- Lactose intolerance.
- Medications (Antibiotics, NSAIDs).
- Toxins (Heavy metals, contaminated food).
3. Risk Factors:
- Poor sanitation & hygiene.
- Contaminated food or water.
- Close contact with infected individuals (Daycares, schools).
- Recent travel to endemic areas.
- Weakened immune system (Malnutrition, HIV, immunosuppressive therapy).
Types of Gastroenteritis:
| Type | Cause | Characteristics |
|---|---|---|
| Viral Gastroenteritis | Rotavirus, Norovirus | Watery diarrhea, vomiting, fever, self-limiting |
| Bacterial Gastroenteritis | Salmonella, Shigella, E. coli | Severe diarrhea (bloody in dysentery), fever, abdominal pain |
| Parasitic Gastroenteritis | Giardia, Cryptosporidium | Prolonged diarrhea, bloating, weight loss |
| Toxin-Induced Gastroenteritis | Staphylococcus aureus, Bacillus cereus | Sudden vomiting, cramps, resolves quickly |
Pathophysiology:
- Infection or toxin exposure damages intestinal lining.
- Increased secretion of fluids and electrolytes into the intestine.
- Impaired absorption of nutrients and water, leading to diarrhea.
- Loss of fluids causes dehydration and electrolyte imbalance.
- Severe cases can lead to hypovolemia, metabolic acidosis, and shock.
Clinical Manifestations (Signs & Symptoms):
1. Mild Cases:
- Watery diarrhea (No blood or mucus).
- Nausea, Vomiting.
- Low-grade fever.
- Mild abdominal cramps.
2. Moderate to Severe Cases:
- Frequent watery or bloody diarrhea.
- High fever (≥38.5°C).
- Severe abdominal pain.
- Signs of dehydration:
- Dry mouth, decreased tears.
- Sunken eyes, sunken fontanelle (in infants).
- Reduced urine output (<6 wet diapers per day).
- Lethargy or irritability.
3. Signs of Severe Dehydration (Medical Emergency):
- Severe lethargy, Weak or absent pulse.
- Hypotension, Cold extremities.
- No urine output (Anuria).
Diagnosis:
1. Clinical Assessment:
- History of diarrhea, vomiting, fever, recent travel, or contaminated food exposure.
- Physical examination (Dehydration assessment – Skin turgor, Capillary refill, Mucous membranes).
2. Laboratory Tests (If Severe or Prolonged Case):
- Stool Culture: If blood/mucus in stool, prolonged diarrhea, or immunocompromised child.
- Stool PCR/Antigen Testing: For Rotavirus, Norovirus, Giardia.
- Blood Tests: If dehydration or complications suspected.
- Serum electrolytes (Sodium, Potassium, Bicarbonate).
- CBC (Infection, Hemolytic Uremic Syndrome – HUS in E. coli cases).
- Urinalysis: To assess hydration status.
Medical Management (Treatment):
1. Hydration Therapy (Most Important Treatment)
- Oral Rehydration Therapy (ORT) (Mild-Moderate Dehydration)
- Oral Rehydration Solution (ORS) (WHO formula):
- 50-100 mL/kg over 3-4 hours.
- Small, frequent sips to avoid vomiting.
- Continue breastfeeding or formula feeding in infants.
- Oral Rehydration Solution (ORS) (WHO formula):
- Intravenous Fluids (Severe Dehydration or Shock)
- Ringer’s Lactate or Normal Saline Bolus (20 mL/kg IV over 15-30 min).
- Dextrose-containing IV fluids (If hypoglycemia is present).
2. Dietary Management
- Early refeeding:
- BRAT diet (Banana, Rice, Applesauce, Toast) is no longer recommended.
- Instead, provide easily digestible foods (Cereal, Yogurt, Fruits, Vegetables).
- Avoid sugary drinks, carbonated sodas, and fatty foods.
3. Medications (If Required)
- Antiemetics (Ondansetron) for severe vomiting.
- Antibiotics (For Bacterial Gastroenteritis only):
- Shigella → Azithromycin or Ciprofloxacin.
- C. difficile → Metronidazole or Vancomycin.
- Giardia → Metronidazole (5-7 days).
- Probiotics: Helps in restoring gut flora.
- Zinc supplementation (10-20 mg/day for 10-14 days) → Reduces severity of diarrhea.
Surgical Management:
- Rarely required unless complications develop (e.g., intestinal perforation or volvulus in severe cases).
- For severe Hemolytic Uremic Syndrome (HUS), dialysis may be needed.
Nursing Management:
1. Assessment & Monitoring
- Monitor for dehydration (Urine output, Skin turgor, Vital signs).
- Assess for signs of electrolyte imbalance (Muscle cramps, Weakness, Lethargy).
- Monitor stool frequency and consistency.
2. Infection Control & Prevention
- Hand hygiene (Proper washing before and after patient contact).
- Isolate patients with infectious gastroenteritis (Prevent nosocomial spread).
- Educate parents on safe food handling & water sanitation.
3. Administering Fluids & Medications
- Encourage small, frequent ORS intake.
- Monitor IV fluids carefully (Prevent fluid overload).
- Administer antiemetics and antibiotics as prescribed.
4. Nutritional Support
- Encourage breastfeeding for infants.
- Introduce small, frequent meals post-rehydration.
5. Parental Education
- Teach parents how to prepare ORS at home.
- Educate on early signs of dehydration.
- Advise on vaccination (Rotavirus vaccine to prevent severe cases).
Complications of Gastroenteritis:
- Severe dehydration & hypovolemic shock.
- Electrolyte imbalance (Hypokalemia, Acidosis).
- Hemolytic Uremic Syndrome (HUS) (Shiga-toxin-producing E. coli).
- Malnutrition (Prolonged diarrhea).
- Sepsis (If bacterial infection spreads).
Prognosis:
- Mild cases resolve in 3-7 days with supportive care.
- Severe cases require hospitalization but have a good prognosis if treated promptly.
- Preventable with good hygiene, safe drinking water, and vaccination.
Key Points:
✔ Gastroenteritis is a common childhood illness caused by viral, bacterial, or parasitic infections.
✔ Diarrhea, vomiting, and dehydration are key symptoms.
✔ Oral rehydration therapy (ORT) is the mainstay of treatment.
✔ Antibiotics are only needed for bacterial infections.
✔ Rotavirus vaccination and hygiene practices help prevent gastroenteritis.
Diarrhea in Children
Definition:
Diarrhea is defined as the passage of three or more loose or watery stools per day, or an increase in stool frequency and liquidity compared to the child’s usual pattern. It can lead to dehydration, electrolyte imbalances, and malnutrition, especially in young children.
Etiology (Causes & Risk Factors):
1. Infectious Causes (Most Common)
- Viral Infections:
- Rotavirus (Most common cause worldwide).
- Norovirus (Highly contagious, outbreaks in schools/daycares).
- Adenovirus, Astrovirus (Mild watery diarrhea).
- Bacterial Infections:
- Escherichia coli (E. coli) – Traveler’s diarrhea, Hemolytic Uremic Syndrome (HUS).
- Salmonella – Foodborne, bloody diarrhea.
- Shigella – Dysentery (Fever, mucus, and blood in stools).
- Campylobacter – From contaminated poultry.
- Parasitic Infections:
- Giardia lamblia (Prolonged diarrhea, bloating).
- Cryptosporidium (Common in immunocompromised children).
2. Non-Infectious Causes
- Food intolerance (Lactose intolerance, Cow’s milk protein allergy).
- Malabsorption syndromes (Celiac disease, Cystic fibrosis).
- Inflammatory Bowel Disease (Crohn’s Disease, Ulcerative Colitis).
- Medications (Antibiotics, Laxatives, NSAIDs).
- Irritable Bowel Syndrome (IBS).
3. Risk Factors
- Poor sanitation and contaminated food or water.
- Lack of hand hygiene.
- Close contact with infected individuals (Daycare centers, schools).
- Recent travel to endemic areas.
- Immunosuppression (HIV, Malnutrition).
Types of Diarrhea
| Type | Cause | Characteristics |
|---|---|---|
| Acute Watery Diarrhea | Viral (Rotavirus, Norovirus), Bacterial (E. coli) | Lasts <14 days, causes dehydration |
| Acute Bloody Diarrhea (Dysentery) | Shigella, E. coli (EHEC), Salmonella | Blood/mucus in stools, fever, abdominal pain |
| Persistent Diarrhea | Giardia, Cryptosporidium, Malabsorption syndromes | Lasts >14 days, causes malnutrition |
| Chronic Diarrhea | Celiac disease, Lactose intolerance, IBS | Intermittent, long-term diarrhea with weight loss |
Pathophysiology:
- Infection or inflammation damages the intestinal lining.
- Increased secretion of fluids and electrolytes into the gut lumen.
- Reduced absorption of water and nutrients, leading to diarrhea.
- In severe cases, dehydration, electrolyte imbalance, and malnutrition develop.
Clinical Manifestations (Signs & Symptoms):
1. Mild to Moderate Diarrhea:
- Frequent loose or watery stools.
- Mild abdominal cramps.
- Low-grade fever.
- Mild dehydration (Thirst, Slightly dry mouth).
2. Severe Diarrhea:
- Frequent watery or bloody stools.
- Severe abdominal pain, vomiting.
- High fever (≥38.5°C).
- Signs of dehydration:
- Sunken eyes, dry mouth.
- Reduced urine output (<6 wet diapers/day).
- Irritability, lethargy.
3. Life-Threatening Dehydration (Medical Emergency):
- Lethargy, Weak pulse, Hypotension.
- No urine output (Anuria).
- Cold extremities, Shock.
Diagnosis:
1. Clinical Assessment:
- History: Stool frequency, duration, presence of blood/mucus, recent travel or antibiotic use.
- Physical examination: Signs of dehydration, weight loss, abdominal distension.
2. Laboratory Tests (If Severe or Prolonged Case):
- Stool Culture: If blood/mucus in stool, prolonged diarrhea, or immunocompromised child.
- Stool Microscopy: Detects ova, parasites (Giardia, Cryptosporidium).
- Stool pH & Reducing Substances: If lactose intolerance is suspected.
- Blood Tests:
- Serum electrolytes (Sodium, Potassium, Bicarbonate) for dehydration.
- CBC (For infection, anemia in chronic cases).
- Blood culture (If septicemia suspected).
- Urinalysis: To assess hydration status.
Medical Management (Treatment):
1. Hydration Therapy (Most Important Treatment)
- Oral Rehydration Therapy (ORT) (Mild-Moderate Dehydration)
- ORS (WHO formula):
- 50-100 mL/kg over 3-4 hours.
- Small, frequent sips to avoid vomiting.
- Continue breastfeeding or formula feeding in infants.
- ORS (WHO formula):
- Intravenous Fluids (Severe Dehydration or Shock)
- Ringer’s Lactate or Normal Saline Bolus (20 mL/kg IV over 15-30 min).
- Dextrose-containing IV fluids if hypoglycemia is present.
2. Dietary Management
- Early Refeeding:
- Avoid fasting; continue breastfeeding/formula feeding.
- Easily digestible foods (Rice, banana, yogurt, vegetables, cereals).
- Avoid sugary drinks, carbonated sodas, and fatty foods.
3. Medications (If Required)
- Antiemetics (Ondansetron) for severe vomiting.
- Antibiotics (For Bacterial Diarrhea only):
- Shigella → Azithromycin or Ciprofloxacin.
- Salmonella (Severe cases) → Ceftriaxone.
- C. difficile → Metronidazole or Vancomycin.
- Giardia → Metronidazole (5-7 days).
- Zinc supplementation (10-20 mg/day for 10-14 days) → Reduces severity of diarrhea.
- Probiotics: Helps in restoring gut flora.
Surgical Management:
- Rarely required unless complications develop (e.g., severe intestinal obstruction, volvulus, perforation).
- For severe Hemolytic Uremic Syndrome (HUS) from E. coli infections, dialysis may be needed.
Nursing Management:
1. Assessment & Monitoring
- Monitor for dehydration (Urine output, Skin turgor, Capillary refill, Mucous membranes).
- Assess for signs of electrolyte imbalance (Muscle cramps, Weakness, Lethargy).
- Monitor stool frequency, consistency, and signs of blood/mucus.
2. Infection Control & Prevention
- Hand hygiene (Proper washing before and after patient contact).
- Isolate patients with infectious diarrhea (Prevent nosocomial spread).
- Educate parents on food safety, hygiene, and clean water use.
3. Administering Fluids & Medications
- Encourage small, frequent ORS intake.
- Monitor IV fluids carefully (Prevent fluid overload).
- Administer antiemetics and antibiotics as prescribed.
4. Nutritional Support
- Encourage breastfeeding for infants.
- Introduce small, frequent meals post-rehydration.
5. Parental Education
- Teach parents how to prepare ORS at home.
- Educate on early signs of dehydration.
- Advise on vaccination (Rotavirus vaccine to prevent severe cases).
Complications of Diarrhea:
- Severe dehydration & hypovolemic shock.
- Electrolyte imbalance (Hypokalemia, Acidosis).
- Hemolytic Uremic Syndrome (HUS, from E. coli).
- Malnutrition (Prolonged diarrhea).
- Sepsis (If bacterial infection spreads).
Prognosis:
- Mild cases resolve in 3-7 days with supportive care.
- Severe cases require hospitalization but have a good prognosis if treated promptly.
- Preventable with good hygiene, safe drinking water, and vaccination.
Key Points:
✔ Diarrhea in children is mostly caused by infections (viral, bacterial, or parasitic).
✔ Dehydration is the main risk; ORS is the first-line treatment.
✔ Antibiotics are only needed for bacterial diarrhea.
✔ Hygiene practices and vaccination help prevent diarrhea
Vomiting in Children

Definition:
Vomiting is the forceful expulsion of stomach contents through the mouth due to involuntary contraction of the abdominal muscles and diaphragm. It is a symptom, not a disease, and can be caused by gastrointestinal, neurological, metabolic, or infectious conditions.
Etiology (Causes & Risk Factors):
1. Gastrointestinal Causes (Most Common)
- Gastroenteritis (Viral, Bacterial, Parasitic Infections)
- Gastroesophageal Reflux Disease (GERD)
- Pyloric Stenosis (Projectile vomiting in infants)
- Intussusception (Bilious vomiting, Currant jelly stools)
- Malrotation with volvulus (Medical emergency, Bilious vomiting)
- Food allergies (Milk, Soy, Egg intolerance in infants)
2. Neurological Causes
- Increased Intracranial Pressure (Meningitis, Hydrocephalus, Brain tumor)
- Migraine headaches (Cyclic vomiting syndrome)
- Vestibular disorders (Motion sickness, Labyrinthitis)
3. Metabolic & Endocrine Causes
- Diabetic ketoacidosis (DKA, Fruity breath, Dehydration)
- Congenital adrenal hyperplasia (Salt-wasting crisis)
- Uremia (Kidney failure, Toxin buildup)
- Inborn errors of metabolism (Phenylketonuria, Galactosemia)
4. Toxic Causes
- Food poisoning (Staphylococcus, Bacillus cereus, Clostridium botulinum)
- Drug-induced (Chemotherapy, Antibiotics, NSAIDs, Iron overdose)
- Lead poisoning (Chronic vomiting, Behavioral changes)
5. Psychological Causes
- Anxiety, Stress-related vomiting (School phobia, Emotional distress)
- Bulimia nervosa (Self-induced vomiting in adolescents)
Types of Vomiting:
| Type | Characteristics | Common Causes |
|---|---|---|
| Non-Bilious Vomiting | Stomach contents without bile | GERD, Pyloric stenosis, Overfeeding |
| Bilious Vomiting | Greenish bile-stained vomit | Intestinal obstruction (Malrotation, Volvulus, Intussusception) |
| Projectile Vomiting | Sudden forceful vomiting without nausea | Pyloric stenosis, Brain tumor |
| Cyclic Vomiting Syndrome | Recurrent episodes of severe vomiting | Migraines, Metabolic disorders |
| Hematemesis (Bloody Vomit) | Vomiting with blood | Gastritis, Esophageal varices, Ingestion of blood (Nosebleed) |
Pathophysiology:
- Vomiting center in the brainstem (Medulla Oblongata) is activated by:
- Gastrointestinal irritation (Vagal nerve stimulation).
- Toxins, drugs, or metabolic imbalances (Chemoreceptor trigger zone – CTZ).
- Motion sickness or vestibular disturbances (Inner ear stimulation).
- Emotional stress (Cortical inputs).
- Diaphragm and abdominal muscles contract → Increases intra-abdominal pressure.
- Lower esophageal sphincter relaxes → Stomach contents are expelled.
Clinical Manifestations (Signs & Symptoms):
1. Mild to Moderate Vomiting:
- Intermittent vomiting, No dehydration.
- No significant weight loss.
- Normal activity levels.
2. Severe or Persistent Vomiting:
- Frequent vomiting leading to dehydration (Dry mouth, Sunken eyes, No tears).
- Weight loss, Lethargy.
- Severe abdominal pain (Suggests obstruction, appendicitis).
- High fever (Suggests infection like gastroenteritis).
3. Emergency Signs (Life-Threatening Conditions):
- Bilious (Green) Vomiting → Suggests intestinal obstruction (Volvulus, Intussusception).
- Projectile Vomiting → Suggests Pyloric Stenosis, Brain Tumor.
- Blood in Vomit (Hematemesis) → Suggests GI bleeding, Esophageal varices.
- Altered Consciousness, Seizures → Suggests Increased Intracranial Pressure.
Diagnosis:
1. Clinical History & Examination:
- Onset, Frequency, Relation to meals.
- Color of vomit (Bilious, Bloody, Coffee-ground).
- Associated symptoms (Diarrhea, Fever, Abdominal pain, Headache).
- Recent food intake, Travel history, Medication use.
2. Laboratory Tests (For Severe or Persistent Cases):
- Blood Tests:
- Serum Electrolytes (Na, K, Cl, HCO₃) – Check for dehydration, metabolic alkalosis.
- Glucose (Hypoglycemia in metabolic disorders).
- CBC (Signs of infection or anemia).
- Liver & Kidney Function Tests (Hepatic or renal cause).
- Urinalysis:
- Ketones (Diabetic ketoacidosis, Starvation).
- Urinary sodium (Salt-wasting conditions).
- Stool Culture: If associated with diarrhea (Bacterial, Parasitic infections).
3. Imaging Studies:
- Abdominal Ultrasound (Pyloric stenosis, Intussusception).
- X-ray or Contrast Studies (Malrotation, Volvulus).
- CT or MRI Brain (If neurological symptoms present – Tumor, Hydrocephalus).
Medical Management (Treatment):
1. Hydration Therapy (Most Important)
- Oral Rehydration Therapy (Mild to Moderate Dehydration)
- ORS (WHO formula) – Small frequent sips to prevent further vomiting.
- Continue breastfeeding or formula feeding in infants.
- Intravenous Fluids (Severe Dehydration or Shock)
- Ringer’s Lactate or Normal Saline (Bolus 20 mL/kg IV over 30 minutes).
- Dextrose-containing IV fluids (If hypoglycemia is present).
2. Dietary Management
- Gradual Refeeding:
- Easily digestible foods (Bananas, Rice, Applesauce, Toast – BRAT diet).
- Avoid dairy and fatty foods initially.
3. Medications (If Needed)
- Antiemetics (For Persistent Vomiting)
- Ondansetron (Zofran) – Blocks serotonin in the brainstem (5-HT3 antagonist).
- Metoclopramide – Used in severe cases (Contraindicated in young infants).
- Proton Pump Inhibitors (For GERD-Related Vomiting)
- Omeprazole, Ranitidine.
- Antibiotics (For Bacterial Infections Only)
- Not routinely used unless severe gastroenteritis, sepsis, or H. pylori infection.
- Zinc Supplementation (10-20 mg/day for 10-14 days) → Reduces severity of diarrhea-related vomiting.
Surgical Management:
- Pyloric Stenosis → Pyloromyotomy (Ramstedt’s Procedure).
- Intestinal Obstruction (Malrotation, Volvulus, Intussusception) → Surgical Correction.
- Appendicitis → Appendectomy.
- Increased Intracranial Pressure → Neurosurgical procedures (Shunt placement, Tumor removal).
Nursing Management:
1. Assessment & Monitoring
- Monitor for dehydration (Urine output, Capillary refill, Mucous membranes).
- Check for signs of electrolyte imbalance (Weakness, Muscle cramps, Lethargy).
- Observe for bilious or bloody vomiting (Indicates serious conditions).
2. Infection Control & Prevention
- Hand hygiene, Safe food handling to prevent foodborne illnesses.
- Isolation for infectious causes like gastroenteritis.
3. Administering Fluids & Medications
- Encourage ORS intake, Monitor IV fluids.
- Administer prescribed antiemetics.
4. Parental Education
- Teach parents to recognize signs of dehydration and seek care early.
- Explain dietary modifications post-vomiting.
Complications of Vomiting:
- Dehydration & Hypovolemic Shock.
- Electrolyte Imbalance (Hypokalemia, Metabolic Alkalosis).
- Aspiration Pneumonia (If vomiting while lying down).
- Malnutrition & Weight Loss (Chronic vomiting cases).
Prognosis:
- Most cases resolve with hydration & symptomatic treatment.
- Serious causes require early identification & surgical intervention.
- Good hygiene & vaccination (Rotavirus) can prevent infectious vomiting.
Protein Energy Malnutrition (PEM) in Children
Definition:
Protein Energy Malnutrition (PEM) is a nutritional disorder resulting from a deficiency of proteins, energy (calories), or both, leading to growth retardation, muscle wasting, and impaired immune function. It is more prevalent in developing countries and among children with poor dietary intake, infections, or chronic diseases.
Etiology (Causes & Risk Factors):
1. Primary Causes (Due to Inadequate Intake)
- Insufficient food availability (Poverty, Famine, War).
- Poor weaning practices (Delayed or inadequate introduction of complementary foods).
- Malnutrition in mothers leading to poor fetal growth (Low birth weight, Prematurity).
2. Secondary Causes (Due to Underlying Diseases)
- Chronic infections (Tuberculosis, HIV/AIDS, Measles, Chronic diarrhea).
- Malabsorption disorders (Celiac disease, Cystic fibrosis, Lactose intolerance).
- Congenital anomalies (Cleft lip/palate leading to feeding difficulties).
- Increased metabolic demand (Burns, Cancer, Hyperthyroidism).
3. Risk Factors
- Low socioeconomic status, Poor hygiene & sanitation.
- Frequent infections (Diarrhea, Respiratory infections).
- Lack of exclusive breastfeeding in the first 6 months.
Types of PEM:
| Type | Cause | Clinical Features |
|---|---|---|
| Marasmus (Severe Caloric Deficiency) | Inadequate calorie intake | Severe wasting, No edema, Muscle loss, Wrinkled skin |
| Kwashiorkor (Severe Protein Deficiency) | Low protein intake with normal calories | Edema, Moon face, Skin lesions, Hepatomegaly, Hair discoloration |
| Marasmic-Kwashiorkor (Mixed Type) | Combined protein & calorie deficiency | Features of both Marasmus & Kwashiorkor, High risk of death |
Pathophysiology:
- Inadequate intake of proteins or calories leads to energy depletion.
- Body starts using fat and muscle protein for energy → Muscle wasting & weight loss.
- Loss of plasma proteins (Kwashiorkor) causes:
- Edema due to reduced oncotic pressure.
- Fatty liver due to impaired protein transport.
- Severe immune suppression → Increased risk of infections.
- Malabsorption & Gut atrophy → Chronic diarrhea & poor nutrient absorption.
Clinical Manifestations (Signs & Symptoms):
1. Marasmus (Severe Caloric Deficiency)
- Extreme muscle wasting, No subcutaneous fat.
- Thin, wrinkled skin (“Old man’s face”).
- Severe growth retardation.
- No edema, Irritability, Excessive crying.
2. Kwashiorkor (Severe Protein Deficiency)
- Generalized edema (Swollen legs, face, abdomen).
- “Flaky paint” skin rash, Depigmentation & skin peeling.
- Sparse, brittle, easily pluckable hair (“Flag sign” – alternating bands of pale & normal hair).
- Enlarged liver (Fatty liver due to impaired lipoprotein synthesis).
- Apathy, Lethargy, Poor appetite.
3. Marasmic-Kwashiorkor (Mixed Type)
- Wasting with edema.
- Severe infections, Skin lesions.
- High risk of death due to multi-organ failure.
Diagnosis:
1. Anthropometric Measurements:
- Weight-for-age, Height-for-age (Stunting), Weight-for-height (Wasting).
- Mid-Upper Arm Circumference (MUAC) < 11.5 cm (Severe Malnutrition).
- Body Mass Index (BMI) for age (Severe thinness <3rd percentile).
2. Laboratory Investigations:
- Serum Albumin (Low in Kwashiorkor, Normal in Marasmus).
- Hemoglobin (Iron-deficiency anemia is common).
- Electrolytes (Low potassium, Sodium imbalance due to diarrhea).
- Liver function tests (Elevated transaminases in Kwashiorkor).
- Stool examination (Parasitic infections, Fat malabsorption).
3. Clinical Screening Tools:
- WHO Growth Charts (Z-scores for weight, height, BMI).
- Clinical signs of malnutrition (Edema, Muscle wasting, Hair changes).
Medical Management (Treatment):
1. Immediate Stabilization (Phase 1 – Resuscitation)
- Treatment of Hypoglycemia (Dextrose IV or oral sugar water).
- Rehydration with ORS (WHO ReSoMal – Rehydration Solution for Malnutrition).
- Correction of Electrolytes (Potassium, Magnesium, Zinc).
- Treatment of Infections (Broad-spectrum antibiotics – Ampicillin & Gentamicin).
2. Nutritional Rehabilitation (Phase 2 – Recovery)
- Gradual introduction of small, frequent feeds (F-75 Formula – 75 kcal/100 mL).
- Transition to F-100 (100 kcal/100 mL) once stabilized.
- Micronutrient supplementation (Vitamin A, Iron, Zinc, Folic Acid).
- Encourage breastfeeding and appropriate complementary feeding.
3. Catch-Up Growth (Phase 3 – Long-Term Care)
- High-energy, protein-rich diet (Locally available foods).
- Therapeutic foods (Ready-to-use therapeutic foods – RUTF like Plumpy’Nut).
- Growth monitoring & Immunization.
Surgical Management:
- Not commonly required except in complications like:
- Intestinal obstruction (From severe malnutrition-induced gut atrophy).
- Abscess drainage in severe infections.
- Gastrostomy or Feeding Tube Placement (For persistent severe malnutrition with feeding difficulties).
Nursing Management:
1. Assessment & Monitoring
- Daily weight monitoring (Detect response to feeding).
- Monitor for dehydration & signs of hypoglycemia.
- Assess skin condition (Pressure ulcers, Infections).
- Monitor for signs of refeeding syndrome (Electrolyte shifts causing heart failure).
2. Nutritional Support
- Encourage small, frequent feeds (Avoid overfeeding initially).
- Monitor tolerance to therapeutic milk (F-75, F-100).
- Educate caregivers on home-based feeding & hygiene practices.
3. Infection Prevention
- Hand hygiene & clean feeding practices.
- Ensure proper sanitation (Prevent diarrhea & recurrent infections).
- Administer vaccinations (Measles vaccine to prevent complications).
4. Psychosocial Support
- Promote maternal education on nutrition.
- Encourage skin-to-skin contact & bonding.
- Address underlying socioeconomic issues (Food security, Breastfeeding support).
Complications of PEM:
- Severe infections (Pneumonia, Sepsis, Tuberculosis).
- Hypothermia & Hypoglycemia (Common in Marasmus).
- Heart failure (Refeeding syndrome in rapid refeeding cases).
- Permanent growth retardation & Cognitive impairment.
- High risk of mortality if untreated.
Prognosis:
- With early intervention, survival rate is high.
- Children with chronic malnutrition may suffer long-term cognitive and physical deficits.
- Prevention through proper nutrition, breastfeeding, and food security is key.
Key Points:
✔ PEM includes Marasmus (Severe wasting) and Kwashiorkor (Edema, Hair changes).
✔ Causes include poor diet, chronic infections, and malabsorption disorders.
✔ Treatment focuses on gradual refeeding, electrolyte correction, and infection management.
✔ Nursing care includes monitoring hydration, nutritional support, and infection prevention.
✔ Long-term rehabilitation and community-based nutrition programs are essential for prevention.
Hepatic Diseases in Children
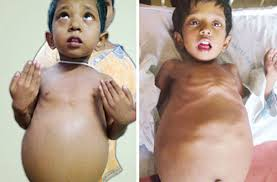
Definition:
Hepatic diseases in children refer to a group of conditions affecting the liver, leading to liver dysfunction, inflammation, infection, metabolic derangements, or structural abnormalities. These diseases can be congenital, infectious, metabolic, autoimmune, or toxic in origin.
Etiology (Causes & Risk Factors):
1. Infectious Causes
- Viral Hepatitis (Hepatitis A, B, C, D, E).
- Bacterial Infections (Sepsis, Typhoid, Tuberculosis).
- Parasitic Infections (Amoebic liver abscess, Hydatid cysts).
2. Metabolic & Genetic Disorders
- Wilson’s Disease (Copper accumulation in the liver).
- Alpha-1 Antitrypsin Deficiency (Genetic cause of liver cirrhosis).
- Glycogen Storage Diseases (Impaired glucose metabolism in the liver).
3. Autoimmune & Inflammatory Conditions
- Autoimmune Hepatitis (Immune system attacks liver cells).
- Primary Sclerosing Cholangitis (Inflammation of bile ducts).
4. Structural & Congenital Defects
- Biliary Atresia (Obstruction or absence of bile ducts).
- Choledochal Cyst (Congenital dilation of bile ducts).
5. Toxic & Drug-Induced Causes
- Drug toxicity (Acetaminophen overdose, Anti-tuberculosis drugs).
- Toxin exposure (Aflatoxin poisoning, Mushroom toxicity).
6. Nutritional Causes
- Non-Alcoholic Fatty Liver Disease (NAFLD) due to obesity and high-fat diet.
- Severe malnutrition leading to hepatic dysfunction (Kwashiorkor).
Types of Pediatric Liver Diseases:
| Type | Cause | Characteristics |
|---|---|---|
| Hepatitis (Acute/Chronic) | Viral, Autoimmune, Toxic | Liver inflammation, Jaundice, Elevated liver enzymes |
| Liver Cirrhosis | Chronic liver disease (Wilson’s, Biliary Atresia, Hepatitis) | Fibrosis, Portal hypertension, Ascites |
| Biliary Atresia | Congenital biliary obstruction | Jaundice in newborns, Clay-colored stools |
| Metabolic Liver Diseases | Wilson’s Disease, Glycogen Storage Disorders | Hepatomegaly, Neurological symptoms |
| Hepatic Tumors | Hepatoblastoma, Hepatocellular Carcinoma | Abdominal mass, Weight loss |
| Neonatal Cholestasis | Impaired bile flow in neonates | Persistent jaundice, Dark urine, Acholic stools |
Pathophysiology:
- Liver Damage or Dysfunction occurs due to infection, toxins, metabolic errors, or immune-mediated destruction.
- Inflammation and Cellular Injury lead to:
- Hepatic necrosis and fibrosis.
- Jaundice due to bilirubin accumulation.
- Coagulopathy due to impaired clotting factor synthesis.
- Impaired Bile Flow (Cholestasis) leads to:
- Fat malabsorption → Vitamin deficiencies (A, D, E, K).
- Clay-colored stools, Dark urine, Pruritus (Itching).
- Progression to Cirrhosis results in:
- Portal hypertension → Ascites, Splenomegaly, Varices.
- Hepatic Encephalopathy → Confusion, Lethargy, Coma.
Clinical Manifestations (Signs & Symptoms):
1. General Symptoms of Liver Disease
- Jaundice (Yellowing of skin & eyes).
- Hepatomegaly (Enlarged liver, Right upper quadrant pain).
- Dark urine, Pale or clay-colored stools (Due to cholestasis).
- Pruritus (Severe itching from bile salt accumulation).
- Poor weight gain, Malnutrition (Due to fat malabsorption).
2. Specific Symptoms Based on Disease Type
| Disease | Key Symptoms |
|---|---|
| Acute Hepatitis | Fever, Fatigue, Nausea, Vomiting, Jaundice, Elevated liver enzymes |
| Chronic Liver Disease (Cirrhosis) | Portal hypertension, Ascites, Varices, Growth failure |
| Biliary Atresia | Persistent jaundice in newborns, Dark urine, Clay-colored stools |
| Wilson’s Disease | Kayser-Fleischer rings (Brown corneal deposits), Neurological symptoms |
| Autoimmune Hepatitis | Hepatomegaly, Fatigue, Joint pain, Liver failure |
| Hepatic Tumors | Palpable abdominal mass, Weight loss, Elevated alpha-fetoprotein (AFP) |
Diagnosis:
1. Blood Tests:
- Liver Function Tests (LFTs):
- Elevated ALT, AST, ALP (Liver enzyme elevation).
- High bilirubin (Direct & Indirect) – Indicates cholestasis.
- Low albumin & Prolonged PT/INR – Suggests liver failure.
- Viral Markers:
- Hepatitis A, B, C, D, E serology.
- Metabolic Tests:
- Serum Copper & Ceruloplasmin (For Wilson’s Disease).
- Blood Ammonia Levels (For Hepatic Encephalopathy).
2. Imaging Studies:
- Abdominal Ultrasound: Evaluates liver size, Cirrhosis, Tumors.
- Hepatobiliary Scan (HIDA Scan): Assesses bile duct patency (For Biliary Atresia).
- CT Scan / MRI: Detects tumors, Fibrosis, Hepatic vascular abnormalities.
3. Liver Biopsy (Definitive Diagnosis for Chronic Liver Disease):
- Identifies inflammation, fibrosis, cirrhosis, or metabolic disorders.
Medical Management (Non-Surgical Treatment):
1. Supportive Therapy:
- Vitamin Supplementation (A, D, E, K) for fat-soluble vitamin deficiency.
- High-calorie, Low-fat Diet with Medium-Chain Triglycerides (MCTs).
- Avoid Hepatotoxic Drugs (Acetaminophen, NSAIDs).
2. Specific Treatments:
- Antiviral Therapy (For Chronic Hepatitis B, C).
- Copper Chelators (For Wilson’s Disease – Penicillamine, Zinc).
- Corticosteroids & Immunosuppressants (For Autoimmune Hepatitis).
- Ursodeoxycholic Acid (For Cholestasis – Improves bile flow).
3. Management of Complications:
- Lactulose for Hepatic Encephalopathy.
- Diuretics (Spironolactone, Furosemide) for Ascites.
- Propranolol for Portal Hypertension.
Surgical Management:
1. Biliary Atresia – Kasai Procedure (Hepatoportoenterostomy)
- Restores bile drainage by connecting the liver to the small intestine.
2. Liver Transplantation (For End-Stage Liver Disease)
- Indicated in:
- Biliary Atresia (Failed Kasai Surgery).
- Liver Cirrhosis with Decompensation.
- Metabolic Liver Diseases (Unresponsive to medical therapy).
Nursing Management:
1. Monitoring & Assessment
- Monitor Liver Function Tests, Bilirubin levels, Ammonia levels.
- Assess for Jaundice, Abdominal Distension, Growth Retardation.
- Observe for Bleeding Tendencies (Coagulopathy in Liver Failure).
2. Nutritional Support
- Encourage high-calorie, high-protein diet.
- Monitor for signs of malnutrition (Muscle wasting, Poor growth).
3. Infection Prevention
- Strict hand hygiene to prevent infections.
- Ensure timely vaccinations (Hepatitis B, A).
4. Parent & Family Education
- Explain the importance of long-term follow-up & medication adherence.
- Educate about early signs of liver failure (Confusion, Ascites, GI Bleeding).
Complications of Pediatric Liver Diseases:
- Liver Cirrhosis → Portal Hypertension, Ascites, Varices.
- Hepatic Encephalopathy → Confusion, Coma.
- Liver Failure → Coagulopathy, Hypoglycemia.
- Hepatocellular Carcinoma (Chronic Hepatitis, Wilson’s Disease).
Prognosis:
- Early diagnosis & treatment improve outcomes.
- Biliary Atresia untreated → Liver failure by age 2-3.
- Liver Transplantation is the definitive cure for end-stage liver disease.
Key Points:
✔ Hepatic diseases in children can be congenital, infectious, metabolic, or autoimmune.
✔ Common symptoms include jaundice, hepatomegaly, pruritus, and ascites.
✔ Treatment depends on the cause – antiviral therapy, copper chelators, steroids, or surgery.
✔ Liver transplantation is the definitive treatment for end-stage liver disease.
Intestinal Parasites in Children
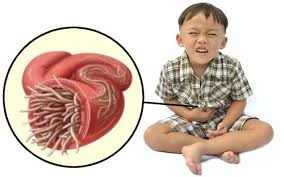
Definition:
Intestinal parasites are organisms that live in the intestines of a child and derive nutrients at the host’s expense. These can be protozoa (single-celled organisms) or helminths (worms) and cause malnutrition, diarrhea, anemia, and developmental delays.
Etiology (Causes & Risk Factors):
1. Infectious Agents (Types of Parasites)
A. Protozoa (Microscopic, Single-Celled Parasites)
- Giardia lamblia → Causes Giardiasis (Chronic diarrhea, Malabsorption).
- Entamoeba histolytica → Causes Amebiasis (Bloody diarrhea, Liver abscess).
- Cryptosporidium → Causes Cryptosporidiosis (Severe diarrhea, Common in immunocompromised children).
B. Helminths (Worms – Multicellular Parasites)
- Roundworms (Nematodes)
- Ascaris lumbricoides (Ascariasis – Intestinal blockage, Poor growth).
- Enterobius vermicularis (Pinworms – Perianal itching).
- Strongyloides stercoralis (Strongyloidiasis – Persistent diarrhea, Lung migration).
- Flatworms (Cestodes – Tapeworms)
- Taenia species (Tapeworm infection – Malnutrition, Abdominal pain).
- Hymenolepis nana (Dwarf tapeworm – Common in children).
- Flukes (Trematodes)
- Schistosoma species (Schistosomiasis – Chronic anemia, Growth retardation).
2. Risk Factors:
- Poor sanitation & contaminated drinking water.
- Consumption of raw or undercooked meat/fish.
- Barefoot walking (Hookworm infection from soil).
- Poor hygiene (Lack of handwashing).
- Crowded living conditions, Poor waste disposal.
- Immunocompromised children (HIV/AIDS, Malnourished).
Types of Intestinal Parasite Infections:
| Type | Parasite | Common Symptoms |
|---|---|---|
| Protozoal Infections | Giardia, Amoeba, Cryptosporidium | Diarrhea, Malabsorption, Dehydration |
| Roundworms (Nematodes) | Ascaris, Hookworms, Strongyloides | Abdominal pain, Intestinal obstruction, Anemia |
| Pinworms (Enterobius) | Enterobius vermicularis | Perianal itching, Sleep disturbance |
| Tapeworms (Cestodes) | Taenia, Hymenolepis | Weight loss, Malnutrition, Vitamin B12 deficiency |
| Flukes (Trematodes) | Schistosoma | Chronic anemia, Liver fibrosis |
Pathophysiology:
- Ingestion or penetration of parasite eggs/larvae occurs through contaminated food, water, soil, or direct skin contact.
- Parasites mature in the intestine and disrupt normal nutrient absorption.
- Effects on the host include:
- Malnutrition (Nutrient depletion by the parasite).
- Intestinal damage & inflammation (Diarrhea, Ulcers).
- Migration to other organs (Liver, Lungs, Brain – Depending on parasite type).
- Chronic infections lead to anemia, stunted growth, cognitive impairment.
Clinical Manifestations (Signs & Symptoms):
1. General Symptoms Common to Most Intestinal Parasites
- Diarrhea (Watery, Chronic, or Bloody).
- Abdominal pain, Bloating, Cramping.
- Weight loss, Growth retardation.
- Loss of appetite, Fatigue.
- Malabsorption (Vitamin & Iron Deficiency → Anemia).
- Nausea, Vomiting.
2. Specific Symptoms Based on Parasite Type
| Parasite | Key Symptoms |
|---|---|
| Giardia (Giardiasis) | Chronic watery diarrhea, Foul-smelling stool, Fatigue, Bloating |
| Entamoeba histolytica (Amebiasis) | Bloody diarrhea, Liver abscess (Right upper quadrant pain, Fever) |
| Ascaris (Roundworm) | Large worm in stool/vomit, Intestinal blockage, Pneumonia (Lung migration) |
| Enterobius (Pinworms) | Perianal itching (Worse at night), Sleep disturbances |
| Hookworms (Necator, Ancylostoma) | Chronic anemia, Weakness, Pica (Eating non-food items) |
| Tapeworms (Taenia, Hymenolepis) | Weight loss, Vitamin B12 deficiency, Neurological symptoms (If larvae reach brain) |
Diagnosis:
1. Laboratory Tests:
- Stool Examination (Microscopy, Ova & Parasite Test): Detects eggs, larvae, cysts of parasites.
- Stool Antigen Tests (For Giardia, Cryptosporidium).
- Blood Tests:
- Eosinophilia (Increased in Helminth infections).
- Serologic Tests (For Amoebiasis, Schistosomiasis).
- Fecal Occult Blood Test (FOBT): Detects hidden blood (For Amoebiasis, Hookworms).
2. Imaging Studies:
- Ultrasound (For Hepatic Amoebiasis, Hydatid Cysts).
- X-ray/CT Scan (For Ascaris Intestinal Blockage, Neurocysticercosis).
Medical Management (Treatment):
1. Anti-Parasitic Medications
| Parasite | Drug of Choice |
|---|---|
| Giardia lamblia | Metronidazole, Tinidazole |
| Entamoeba histolytica | Metronidazole + Paromomycin |
| Ascaris (Roundworm), Hookworm, Pinworm | Albendazole, Mebendazole |
| Tapeworms (Taenia, Hymenolepis) | Praziquantel, Niclosamide |
| Strongyloides | Ivermectin |
| Schistosoma (Flukes) | Praziquantel |
2. Symptomatic Treatment:
- Rehydration (ORS, IV Fluids for Severe Dehydration).
- Iron & Vitamin Supplementation (For Anemia & Malnutrition).
- Probiotics (Restore gut flora post-treatment).
Surgical Management:
- Rarely required unless complications occur:
- Intestinal obstruction due to Ascaris (Surgical removal).
- Liver abscess from Amoebiasis (Drainage required if large).
- Hydatid cyst (Surgical excision if causing pressure effects).
Nursing Management:
1. Prevention & Hygiene Education:
- Encourage handwashing after defecation & before eating.
- Promote drinking boiled or filtered water.
- Proper food handling & cooking meat thoroughly.
- Avoid walking barefoot in contaminated soil.
- Regular deworming in endemic areas (WHO recommends every 6 months).
2. Assessment & Monitoring:
- Monitor for signs of dehydration, Malnutrition, Growth delays.
- Check for stool consistency, Blood/mucus in stools.
- Assess for perianal itching (Suggests Pinworms).
3. Medication Administration:
- Ensure completion of anti-parasitic treatment.
- Monitor for side effects of drugs (Nausea, Dizziness, Liver toxicity).
4. Nutritional Support:
- Encourage a protein-rich diet (To aid recovery).
- Supplement Iron, Vitamin B12 for anemic children.
- Probiotic-rich foods (Yogurt) to restore gut flora post-treatment.
Complications of Intestinal Parasite Infections:
- Severe Malnutrition & Growth Retardation.
- Anemia (Hookworms, Schistosomiasis).
- Intestinal Obstruction (Ascaris, Tapeworms).
- Liver Abscess (Amoebiasis).
- Neurological Symptoms (Neurocysticercosis from Taenia Solium).
Prognosis:
- Good prognosis with early detection & treatment.
- Complications arise in untreated or chronic cases (Anemia, Growth delays).
- Deworming programs significantly reduce infections in endemic regions.
Key Points:
✔ Intestinal parasites are common in children, especially in areas with poor sanitation.
✔ Symptoms include diarrhea, malnutrition, anemia, and growth failure.
✔ Diagnosis is done through stool microscopy, antigen tests, and serology.
✔ Treatment involves anti-parasitic medications, hydration, and nutritional support.
✔ Prevention includes good hygiene, safe water, and periodic deworming
Wilms Tumor (Nephroblastoma) in Children
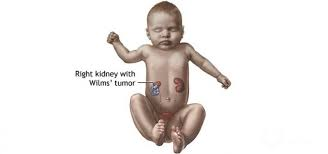
Definition:
Wilms tumor, also known as nephroblastoma, is a malignant tumor of the kidney that primarily affects children. It is the most common renal cancer in pediatric patients and usually occurs in children under the age of 5 years.
Etiology (Causes & Risk Factors):
- Genetic Factors:
- Associated with specific genetic mutations, such as mutations in the WT1 or WT2 genes located on chromosome 11p13.
- Beckwith-Wiedemann syndrome, WAGR syndrome (Wilms tumor, Aniridia, Genitourinary anomalies, and mental Retardation), and Denys-Drash syndrome increase the risk.
- Family History:
- Having a family history of Wilms tumor increases risk.
- Congenital Malformations:
- Children with congenital anomalies, such as aniridia (absence of the iris), hemihypertrophy (one side of the body larger than the other), and cryptorchidism (undescended testes), have a higher risk.
Types of Wilms Tumor:
- Favorable Histology:
- Contains well-differentiated cells and has a better prognosis.
- Unfavorable (Anaplastic) Histology:
- Contains poorly differentiated or anaplastic cells, making it more aggressive and difficult to treat.
Pathophysiology:
- Genetic mutations in the WT1 or WT2 genes lead to abnormal cell growth in the developing kidneys.
- Rapid, uncontrolled proliferation of nephroblastic cells leads to tumor formation.
- The tumor may invade nearby structures, such as the renal vein or inferior vena cava.
- In advanced stages, the tumor may metastasize to other organs, including the lungs, liver, and lymph nodes.
Clinical Manifestations (Signs & Symptoms):
- Abdominal Mass:
- Most common symptom; often painless and felt as a firm, smooth mass on one side of the abdomen.
- Abdominal Pain:
- Due to pressure effects of the growing tumor.
- Hematuria (Blood in urine):
- Occurs in a minority of cases.
- Hypertension:
- Caused by tumor compression on renal blood vessels, leading to activation of the renin-angiotensin system.
- Fever:
- Due to tumor necrosis or secondary infection.
- Weight Loss and Fatigue:
- General symptoms seen in advanced disease.
- Symptoms of Metastasis:
- Cough and shortness of breath if the tumor has spread to the lungs.
Diagnosis:
1. Physical Examination:
- Abdominal palpation may reveal a firm, non-tender mass.
2. Imaging Studies:
- Abdominal Ultrasound:
- Initial imaging to confirm the presence of a renal mass.
- CT Scan or MRI of the Abdomen:
- Provides detailed information about the tumor size, location, and possible spread.
- Chest X-ray or CT Scan of the Chest:
- To evaluate for lung metastasis.
3. Laboratory Tests:
- Urinalysis:
- Detects hematuria.
- Complete Blood Count (CBC):
- May show anemia due to blood loss or tumor burden.
- Liver and Renal Function Tests:
- Assess kidney and liver involvement.
- Coagulation Profile:
- To detect bleeding disorders associated with Wilms tumor.
4. Biopsy:
- Histopathological examination of the tumor tissue is performed if imaging findings are inconclusive.
Medical Management:
1. Chemotherapy:
- Administered preoperatively (neoadjuvant) to shrink the tumor or postoperatively (adjuvant) to destroy remaining cancer cells.
- Commonly used agents include:
- Vincristine
- Dactinomycin
- Doxorubicin
- Chemotherapy regimens are based on the tumor’s histology and stage.
2. Radiation Therapy:
- Used in cases with unfavorable histology or advanced-stage disease.
- Targeted radiation is used to destroy remaining cancer cells after surgery.
Surgical Management:
1. Nephrectomy:
- Radical Nephrectomy:
- Removal of the affected kidney along with the tumor, adrenal gland, and surrounding tissues.
- Partial Nephrectomy:
- Removal of the tumor only, sparing as much healthy kidney tissue as possible (used for bilateral tumors).
2. Surgical Approach:
- Staging Laparotomy:
- To determine the extent of the disease and presence of metastasis.
- Lymph Node Dissection:
- Removal of regional lymph nodes for staging purposes.
Nursing Management:
1. Preoperative Care:
- Assessment:
- Monitor vital signs, including blood pressure (hypertension is common).
- Assess for signs of hematuria, abdominal mass, and pain.
- Psychological Support:
- Provide emotional support to the child and family.
- Educate the family about the surgical procedure and treatment plan.
- Preoperative Preparations:
- Ensure the child is NPO (nothing by mouth) before surgery.
- Administer prescribed preoperative medications.
2. Postoperative Care:
- Monitor Vital Signs:
- Watch for signs of bleeding, infection, and changes in blood pressure.
- Pain Management:
- Administer analgesics as prescribed for pain relief.
- Fluid and Electrolyte Balance:
- Monitor intake and output to ensure proper hydration.
- Wound Care:
- Assess the surgical site for signs of infection or dehiscence.
- Dietary Management:
- Encourage a high-protein, high-calorie diet for recovery.
3. Supportive Care:
- Monitor for Side Effects of Chemotherapy:
- Watch for nausea, vomiting, hair loss, and signs of infection (neutropenia).
- Monitor for Side Effects of Radiation Therapy:
- Assess for skin irritation, fatigue, and potential organ damage.
- Psychosocial Support:
- Provide ongoing emotional support and counseling for the child and family.
- Education:
- Educate parents about signs of recurrence, follow-up care, and coping strategies.
Complications of Wilms Tumor:
- Metastasis:
- Most commonly to the lungs and liver.
- Tumor Rupture:
- Increases the risk of peritoneal seeding and spread of cancer cells.
- Hypertension:
- Due to tumor compression on renal blood vessels.
- Infection and Bleeding:
- Postoperative complications due to surgery.
- Chronic Kidney Disease:
- In cases where one or both kidneys are removed.
Prognosis:
- The prognosis depends on the tumor’s stage, histology, and response to treatment.
- Favorable histology tumors have a 90% survival rate if detected early and treated appropriately.
- Unfavorable histology tumors have a lower survival rate but can be managed with aggressive treatment.
Key Points:
✔ Wilms tumor is the most common pediatric renal cancer, often presenting as an abdominal mass.
✔ Early diagnosis and a combination of surgery, chemotherapy, and radiation offer the best outcomes.
✔ Nursing care focuses on preoperative and postoperative management, pain relief, and monitoring for complications.
✔ Ongoing follow-up is crucial for detecting recurrence and managing late effects of treatment.
Exstrophy of the Bladder in Children
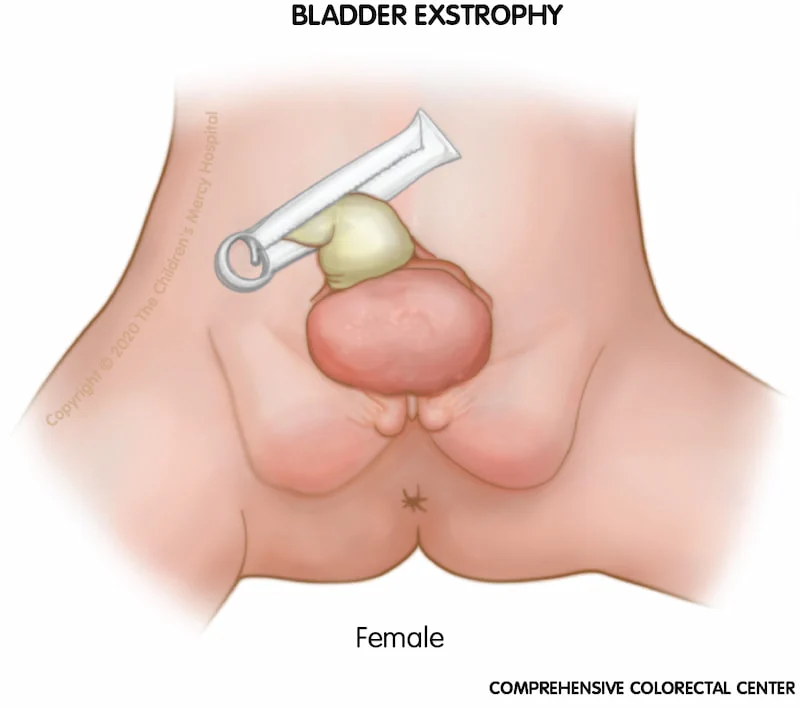
Definition:
Bladder exstrophy is a rare congenital anomaly in which the bladder develops outside the abdominal wall, leading to exposure of the bladder mucosa, urethra, and sometimes the genitalia. It is a part of the bladder-exstrophy-epispadias complex (BEEC) and can be associated with pelvic bone abnormalities.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Family history increases the risk.
- Associated with chromosomal abnormalities (trisomy 18).
2. Developmental Factors:
- Failure of the lower abdominal wall and bladder to close during fetal development.
- Abnormal mesodermal migration during weeks 4-6 of gestation.
3. Environmental Factors:
- Maternal exposure to teratogens (chemicals, medications, radiation).
- Advanced maternal age.
- Use of hormonal medications during pregnancy.
Types of Bladder Exstrophy:
| Type | Description |
|---|---|
| Classic Bladder Exstrophy | Bladder is open and exposed on the abdominal wall. |
| Cloacal Exstrophy | Severe form where bladder, intestines, and genitalia are exposed. |
| Epispadias | Urethral opening is misplaced on the dorsal (upper) side of the penis or clitoris. |
Pathophysiology:
- Failure of the lower abdominal wall and bladder fusion during fetal development.
- Exposure of the bladder mucosa leads to continuous urine leakage.
- Incomplete development of the pelvic bones affects locomotion.
- Risk of recurrent infections, impaired renal function, and infertility if untreated.
Clinical Manifestations (Signs & Symptoms):
1. At Birth:
- Bladder mucosa exposed on the lower abdomen.
- Continuous urine leakage from the exposed bladder.
- Widened pubic bones (Diastasis of the pubic symphysis).
- Short, rotated legs (Pelvic bone abnormalities).
- Umbilicus is low-set and displaced downward.
- Abnormal genitalia (Epispadias, Bifid scrotum in males, Split clitoris in females).
2. Associated Anomalies:
- Hydronephrosis (Swelling of kidneys due to urine buildup).
- Vesicoureteral Reflux (Urine backflow from the bladder to the kidneys).
- Renal insufficiency (Increased risk of kidney failure over time).
Diagnosis:
1. Prenatal Diagnosis (Before Birth)
- Ultrasound (After 16 weeks of gestation):
- Shows absence of a normal bladder.
- Low-set umbilicus, abnormal pubic bones.
- MRI (To confirm the severity of exstrophy).
2. Postnatal Diagnosis (After Birth)
- Physical Examination:
- Clearly visible exposed bladder mucosa.
- Urine leakage.
- X-ray of the Pelvis:
- Confirms pelvic bone abnormalities (Diastasis of the pubic symphysis).
- Renal Ultrasound:
- Evaluates for hydronephrosis or vesicoureteral reflux.
- Voiding Cystourethrogram (VCUG):
- Assesses for urinary reflux and bladder function.
Medical Management (Pre-Surgical Care):
1. Immediate Postnatal Care:
- Protect the exposed bladder with a moist sterile dressing.
- Prevent infection by covering the bladder with plastic wrap.
- Place the infant supine with legs in frog-leg position to reduce tension.
- Monitor for electrolyte imbalances due to continuous urine loss.
- Administer IV fluids to prevent dehydration.
- Prophylactic antibiotics to reduce the risk of infection.
Surgical Management (Definitive Treatment):
Surgical Options:
| Procedure | Purpose | Age |
|---|---|---|
| Stage 1: Initial Bladder Closure | Close the bladder and abdominal wall | Within 48 hours after birth |
| Stage 2: Epispadias Repair | Restore normal urethral function | 6-12 months of age |
| Stage 3: Urinary Continence Surgery | Improve bladder function, prevent leakage | 4-5 years of age |
1. Primary Closure (For Mild Cases)
- Performed within 48 hours after birth.
- Bladder and abdominal wall are surgically closed.
2. Staged Closure (For Severe Cases)
- Bladder closure first, followed by pelvic reconstruction and epispadias repair.
- Urinary diversion (Urostomy) may be done if normal bladder function is not possible.
3. Urinary Continence Surgery
- Performed in older children (4-5 years).
- Procedures include:
- Bladder augmentation (Increases bladder capacity).
- Artificial sphincter placement (Controls urine flow).
Nursing Management:
1. Preoperative Nursing Care:
- Maintain sterility of the exposed bladder with a moist, sterile dressing.
- Prevent infection with antibiotics and frequent dressing changes.
- Monitor hydration status due to urine loss.
- Support parents emotionally and provide education about the condition.
2. Postoperative Nursing Care:
- Position the infant supine with legs in traction (Bryant’s traction) to prevent movement and tension on the surgical site.
- Monitor for signs of infection (Fever, Redness, Swelling).
- Monitor urine output (Signs of urinary retention or obstruction).
- Pain management (Administer analgesics).
- Wound care and dressing changes to prevent infection.
- Encourage feeding and hydration to prevent dehydration.
3. Long-Term Nursing Care:
- Assist in toilet training for urinary continence post-surgery.
- Educate parents on signs of urinary tract infections (UTIs).
- Monitor for delayed growth and development (Common in severe cases).
- Psychosocial support to help the child and family cope with the condition.
Complications of Bladder Exstrophy:
- Urinary Tract Infections (UTIs) – Due to urinary reflux and poor bladder control.
- Renal Impairment – Increased risk of kidney failure.
- Urinary Incontinence – May require multiple surgeries.
- Pelvic & Orthopedic Abnormalities – May lead to mobility issues.
- Psychosocial Challenges – Children may struggle with self-esteem issues.
Prognosis:
- With early surgical intervention, most children can lead normal lives with minor urinary issues.
- Urinary continence may require multiple procedures.
- Lifelong urological monitoring is necessary to prevent kidney damage.
Key Points:
✔ Bladder exstrophy is a rare congenital defect where the bladder develops outside the abdomen.
✔ Immediate postnatal care includes keeping the bladder moist and preventing infection.
✔ Surgical repair is done in multiple stages (Bladder closure, Epispadias repair, Continence surgery).
✔ Nursing care focuses on infection prevention, hydration, pain management, and parental education.
✔ Long-term follow-up is required to ensure bladder function and prevent complications.
Hypospadias in Children
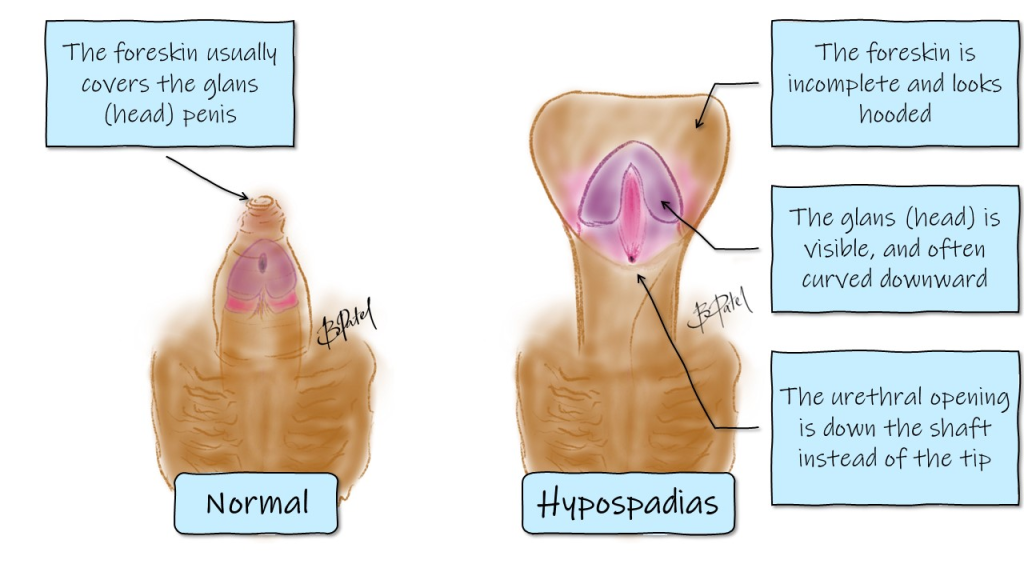
Definition:
Hypospadias is a congenital anomaly in which the urethral opening (meatus) is abnormally positioned on the underside (ventral surface) of the penis, rather than at the tip. It is a common birth defect affecting male infants and may be associated with abnormal foreskin development and penile curvature (chordee).
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Family history of hypospadias.
- Genetic mutations affecting androgen receptor genes.
2. Hormonal & Environmental Factors:
- Defective testosterone production during fetal development.
- Maternal exposure to endocrine disruptors (e.g., pesticides, estrogen-like compounds).
- Advanced maternal age and low birth weight.
3. Associated Conditions:
- Cryptorchidism (Undescended testes).
- Disorders of Sexual Development (DSDs) (e.g., Androgen Insensitivity Syndrome).
Types of Hypospadias (Based on Urethral Opening Location):
| Type | Location of Urethral Opening | Severity |
|---|---|---|
| Anterior (Glanular, Subcoronal) | Near the glans penis | Mild |
| Middle (Midshaft, Penoscrotal) | Along the penile shaft | Moderate |
| Posterior (Scrotal, Perineal) | At the scrotum or perineum | Severe |
Pathophysiology:
- Incomplete fusion of the urethral folds during fetal development (weeks 8-14 of gestation).
- Abnormal positioning of the urethral opening on the ventral side of the penis.
- Incomplete foreskin formation leading to a “hooded” appearance.
- Chordee (Penile curvature) due to fibrous tissue causing downward bending of the penis.
Clinical Manifestations (Signs & Symptoms):
1. Urethral Abnormality:
- Urethral opening is below the normal position (underside of the penis).
- Difficulty urinating while standing (urine stream is directed downward).
2. Foreskin & Penile Abnormalities:
- Hooded foreskin (Incomplete foreskin development).
- Penile curvature (Chordee) may be present in moderate-severe cases.
3. Functional Issues:
- Infertility risk in severe cases (if associated with abnormal sperm delivery).
- Urinary incontinence (Rare but possible).
Diagnosis:
1. Physical Examination:
- Inspection of urethral meatus location.
- Assessment of penile curvature (Chordee test).
- Check for associated genital abnormalities (Cryptorchidism, Micropenis).
2. Imaging Studies (In Severe or Ambiguous Cases):
- Ultrasound of the kidneys & bladder (To rule out urinary tract abnormalities).
- Karyotyping (Genetic Testing) (If disorders of sexual development are suspected).
Medical Management:
**1. Avoid Circumcision at Birth:
- The foreskin is needed for surgical repair later.
- Circumcision is contraindicated in hypospadias.
2. Observation & Hormonal Therapy (For Severe Cases):
- Testosterone therapy (If the penis is underdeveloped or micropenis is present).
Surgical Management (Definitive Treatment):
- Indicated for moderate-severe cases.
- Performed between 6-12 months of age for best outcomes.
Goals of Surgery:
- Relocate the urethral opening to the tip of the penis.
- Correct penile curvature (Chordee).
- Restore normal urinary and reproductive function.
Surgical Procedures:
| Procedure | Indication | Details |
|---|---|---|
| Urethroplasty | All cases requiring correction | Uses tissue grafts (Buccal mucosa or foreskin) to reconstruct the urethra |
| Chordee Correction | If penile curvature is severe | Removes fibrous tissue causing bending |
| One-Stage Repair | Mild to moderate cases | Single surgery to correct meatal position & chordee |
| Two-Stage Repair | Severe or complex cases | Initial correction of chordee followed by urethral reconstruction |
Nursing Management:
1. Preoperative Nursing Care:
- Educate parents about the procedure and expected outcomes.
- Ensure the foreskin is preserved (No circumcision).
- Assess for urinary stream abnormalities (Helps in surgical planning).
- Psychosocial Support:
- Reassure parents about the normalcy of condition.
2. Postoperative Nursing Care:
A. Monitoring & Prevention of Complications:
- Assess urinary output (Monitor for obstruction).
- Monitor for signs of infection (Fever, Redness, Swelling).
- Inspect surgical site for bleeding or wound dehiscence.
B. Pain Management:
- Administer analgesics (Acetaminophen, Ibuprofen).
- Apply topical antibiotics as prescribed.
C. Urinary Catheter Care:
- Post-surgery, a catheter or stent is placed for 1-2 weeks to maintain the new urethral opening.
- Teach parents catheter care to prevent blockages.
D. Preventing Straining & Trauma:
- Prevent constipation (Use stool softeners to avoid straining).
- Avoid rough handling of the penis to prevent surgical complications.
3. Parent Education & Home Care:
- Avoid baths until healing is complete (Use sponge baths instead).
- Monitor for signs of UTI or infection.
- Follow-up visits to assess surgical success.
Complications of Untreated Hypospadias:
- Abnormal Urinary Stream – Causes difficulty urinating while standing.
- Penile Curvature (Chordee) – Can cause pain and erectile dysfunction later.
- Infertility – Severe forms may interfere with sperm delivery.
- Urinary Fistula Formation – Urine leakage from an abnormal opening post-surgery.
- Meatal Stenosis (Narrowed Urethral Opening) – May require further surgery.
Prognosis:
- Excellent outcomes with early surgery.
- Minor cases may not need surgical intervention.
- Children with corrected hypospadias usually have normal urinary and reproductive function.
Key Points:
✔ Hypospadias is a congenital defect where the urethral opening is located on the underside of the penis.
✔ It is classified into anterior, middle, and posterior types based on severity.
✔ Surgical correction (Urethroplasty) is done between 6-12 months of age.
✔ Circumcision should be avoided at birth because the foreskin is used for repair.
✔ Nursing care includes postoperative monitoring, pain management, and catheter care.
Epispadias in Children
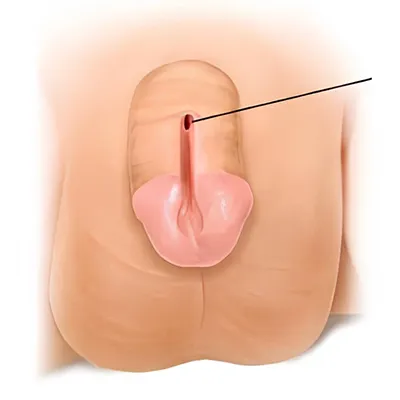
Definition:
Epispadias is a rare congenital anomaly where the urethral opening is located on the dorsal (upper) surface of the penis in males or the clitoris in females. It results from abnormal development of the urethra and external genitalia during fetal development and is often associated with bladder exstrophy.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Familial occurrence suggests genetic predisposition.
- Mutations in genes involved in urogenital development.
2. Developmental Factors:
- Failure of the urethral plate to fuse properly during fetal development (6th–8th week of gestation).
- Deficient mesodermal migration, leading to incomplete closure of the urethra.
3. Associated Conditions:
- Bladder Exstrophy-Epispadias Complex (BEEC).
- Pelvic bone abnormalities (Widened pubic symphysis).
Types of Epispadias:
| Type | Location of Urethral Opening | Features |
|---|---|---|
| Glanular Epispadias | On the glans penis | Mildest form, slight urinary dysfunction |
| Penile Epispadias | Along the penile shaft | Moderate severity, urinary incontinence possible |
| Penopubic Epispadias | Near the bladder neck | Severe form, complete urinary incontinence |
In females:
- The urethral opening is displaced toward the clitoris.
Pathophysiology:
- Defective urethral plate development leads to an open, malformed urethra on the upper surface of the penis or clitoris.
- Bladder and sphincter dysfunction cause urinary incontinence in severe cases.
- Associated widening of the pubic bones further affects urinary and sexual function.
- Risk of recurrent urinary tract infections (UTIs) due to improper urinary drainage.
Clinical Manifestations (Signs & Symptoms):
1. In Males:
- Abnormal dorsal placement of the urethral opening.
- Short, broad, and upward-curving penis (Dorsal chordee).
- Urinary incontinence (Severe cases).
- Difficulty urinating while standing (Spraying of urine).
2. In Females:
- Abnormal urethral opening near the clitoris.
- Short, widened urethra.
- Urinary incontinence (Severe cases).
3. Associated Symptoms:
- Bladder exstrophy in severe cases (Bladder exposed outside the body).
- Widened pubic symphysis affecting walking and posture.
Diagnosis:
1. Physical Examination:
- Inspection of the urethral opening (Located dorsally on the penis or near the clitoris).
- Assessment of urinary control and incontinence severity.
2. Imaging Studies (For Severe Cases):
- Ultrasound of the urinary tract (Checks for associated bladder anomalies).
- Voiding Cystourethrogram (VCUG):
- Assesses bladder function and urinary reflux.
- MRI or CT Scan of the pelvis (For pelvic bone abnormalities).
3. Urodynamic Studies:
- Evaluate bladder capacity, sphincter function, and urine flow rate.
Medical Management (Pre-Surgical Care):
- Prevent urinary infections (Prophylactic antibiotics if needed).
- Encourage fluid intake to reduce urinary stasis.
- Avoid circumcision (Foreskin may be needed for surgical repair).
Surgical Management (Definitive Treatment):
- Surgery is performed between 6-12 months of age for optimal results.
- Goals of surgery:
- Relocate the urethral opening to the normal position.
- Restore urinary continence.
- Correct penile curvature (Chordee correction in males).
- Improve cosmetic appearance of external genitalia.
Surgical Procedures:
| Procedure | Indication | Details |
|---|---|---|
| Modified Cantwell-Ransley Repair | Milder cases | Reconstructs urethra and penis |
| Mitchell Technique | Severe cases | Repositions urethral meatus and reconstructs sphincter |
| Bladder Neck Reconstruction | For incontinence | Strengthens bladder control mechanisms |
| Two-Stage Repair | Complex cases with exstrophy | Initial bladder closure, followed by urethral repair |
Nursing Management:
1. Preoperative Nursing Care:
- Educate parents about the condition, surgery, and postoperative care.
- Monitor for signs of UTIs (Fever, cloudy urine, dysuria).
- Encourage hydration to maintain urinary flow.
2. Postoperative Nursing Care:
A. Monitoring & Infection Prevention:
- Assess urinary output (Monitor catheter function).
- Monitor for surgical site infection (Redness, swelling, pus).
- Administer antibiotics as prescribed.
B. Pain Management:
- Administer analgesics (Acetaminophen, Ibuprofen).
- Apply topical antibiotic ointment if needed.
C. Urinary Catheter Care:
- A catheter or stent is placed for 1-2 weeks post-surgery.
- Teach parents how to care for the catheter at home.
D. Preventing Straining & Trauma:
- Avoid constipation (Give stool softeners if needed).
- Encourage bed rest to reduce strain on the surgical site.
3. Parent Education & Home Care:
- Keep the surgical area clean and dry.
- Monitor for signs of complications (Bleeding, Fever, Pain).
- Avoid rough handling of the penis/clitoris during diaper changes.
- Attend follow-up visits for monitoring urinary function.
Complications of Untreated Epispadias:
- Urinary Incontinence – Severe cases may require multiple surgeries.
- Urinary Reflux (Backflow of urine into kidneys) – Can lead to kidney damage.
- Recurrent Urinary Tract Infections (UTIs).
- Sexual Dysfunction – Possible if not corrected early.
Prognosis:
- Mild cases have an excellent prognosis with early surgery.
- Severe cases may require multiple surgeries but can achieve normal function.
- Bladder function improves significantly with bladder neck reconstruction.
Key Points:
✔ Epispadias is a congenital anomaly where the urethral opening is located on the dorsal (upper) side of the penis or near the clitoris.
✔ It is often associated with bladder exstrophy and urinary incontinence.
✔ Surgical repair (Modified Cantwell-Ransley, Mitchell Technique) is performed between 6-12 months of age.
✔ Nursing care focuses on infection prevention, urinary catheter management, and pain control.
✔ Early surgery improves urinary function, reduces UTIs, and enhances quality of life.
Nephrotic Syndrome in Children
Definition:
Nephrotic Syndrome is a kidney disorder characterized by massive protein loss in urine (proteinuria), low blood protein levels (hypoalbuminemia), swelling (edema), and high cholesterol levels (hyperlipidemia). It results from increased permeability of the glomerular membrane in the kidneys.
Etiology (Causes & Risk Factors):
1. Primary (Idiopathic) Nephrotic Syndrome (Most Common in Children – 90%)
- Minimal Change Disease (MCD) (80% of cases):
- Unknown cause but associated with immune system dysfunction.
- Focal Segmental Glomerulosclerosis (FSGS):
- Scarring (sclerosis) of some parts of the kidney.
- Membranous Nephropathy:
- Thickened glomerular basement membrane.
2. Secondary Nephrotic Syndrome (Due to Other Diseases)
- Autoimmune Diseases (Lupus Nephritis, Henoch-Schönlein Purpura).
- Infections (Hepatitis B, HIV, Malaria).
- Diabetes Mellitus (Diabetic Nephropathy).
- Toxins & Drugs (NSAIDs, Heavy Metals, Allergies).
3. Congenital Nephrotic Syndrome (Rare)
- Genetic mutations affecting kidney function (e.g., Finnish-type nephrotic syndrome).
4. Risk Factors:
- Age: Most common in 2-6 years old.
- Gender: More common in boys than girls.
- Family History: Genetic predisposition increases risk.
Types of Nephrotic Syndrome:
| Type | Cause | Response to Steroids |
|---|---|---|
| Minimal Change Disease (MCD) | Idiopathic (Immune dysfunction) | Good Response |
| Focal Segmental Glomerulosclerosis (FSGS) | Genetic or Secondary Causes | Poor Response |
| Congenital Nephrotic Syndrome | Genetic Mutation | No Response |
Pathophysiology:
- Damage to the glomerular basement membrane increases permeability to proteins.
- Massive protein loss in urine (Proteinuria >3.5g/day).
- Hypoalbuminemia due to excessive protein loss.
- Reduced oncotic pressure → Fluid leaks into tissues → Edema.
- Liver compensates by increasing cholesterol synthesis → Hyperlipidemia.
- Increased risk of infections due to loss of immunoglobulins in urine.
Clinical Manifestations (Signs & Symptoms):
1. Cardinal Features:
- Edema (Puffy eyes, Swollen face, Legs, Abdomen – Ascites).
- Frothy or Foamy Urine (Due to high protein loss).
- Oliguria (Decreased urine output).
- Pallor and Fatigue.
- Weight Gain (Due to fluid retention).
2. Additional Symptoms:
- Hypertension (Rare in MCD but seen in FSGS).
- Abdominal Pain & Anorexia.
- Increased risk of infections (Peritonitis, Pneumonia).
- Thromboembolism (Blood clot formation due to increased fibrinogen).
Diagnosis:
1. Laboratory Tests:
- Urinalysis:
- Massive proteinuria (>3.5g/day).
- No hematuria (if present, consider FSGS).
- Blood Tests:
- Low serum albumin (<2.5g/dL).
- High cholesterol (Hyperlipidemia).
- Normal kidney function (In MCD, altered in secondary nephrotic syndrome).
- 24-Hour Urine Protein Test: Confirms nephrotic-range proteinuria.
2. Imaging Studies:
- Renal Ultrasound: To rule out structural abnormalities.
3. Kidney Biopsy:
- Indicated in steroid-resistant cases (FSGS, Congenital Nephrotic Syndrome).
Medical Management (Non-Surgical Treatment):
1. Corticosteroids (First-Line Therapy)
- Prednisolone (2 mg/kg/day for 6-8 weeks).
- Good response in Minimal Change Disease (MCD).
- Slow tapering to prevent relapse.
2. Immunosuppressants (For Steroid-Resistant Cases)
- Cyclophosphamide or Cyclosporine (For FSGS).
- Rituximab (Monoclonal Antibody Therapy in Severe Cases).
3. Diuretics (For Severe Edema)
- Furosemide (Lasix) with IV Albumin Infusion to reduce fluid overload.
4. Antihypertensive & Anticoagulants
- ACE Inhibitors (Enalapril, Captopril) → Reduces protein loss.
- Aspirin or Heparin → Prevents thrombosis in severe cases.
5. Infection Prevention
- Pneumococcal & Influenza Vaccination (High infection risk).
- Antibiotic therapy for infections.
6. Nutritional Management
- High-protein diet (If protein loss is excessive).
- Low-sodium diet (To prevent worsening edema).
Surgical Management:
- Bilateral Nephrectomy (Last Resort for Congenital Nephrotic Syndrome).
- Kidney Transplant (For End-Stage Kidney Disease – ESRD).
Nursing Management:
1. Pre-Treatment Nursing Care:
- Monitor vital signs (BP, Heart rate, Respiratory rate).
- Assess for edema (Daily weight, Abdominal girth measurement).
- Strict intake-output monitoring (Oliguria, Fluid retention).
- Encourage high-protein, low-sodium diet.
2. Nursing Care During Treatment:
A. Medication Administration
- Administer corticosteroids & immunosuppressants as prescribed.
- Monitor for side effects (Steroid-induced hyperglycemia, Cushing’s Syndrome).
B. Monitoring Fluid Balance
- Measure urine output hourly (Oliguria risk).
- Monitor for signs of fluid overload (Pulmonary edema, Hypertension).
C. Infection Prevention
- Hand hygiene and infection control.
- Administer vaccines as per schedule.
- Encourage parents to avoid crowded areas during treatment.
D. Managing Edema
- Elevate swollen limbs to reduce swelling.
- Encourage mobility to prevent blood clots.
E. Psychosocial Support
- Counsel parents on disease course & treatment expectations.
- Encourage school attendance when possible.
Complications of Nephrotic Syndrome:
- Severe Infections (Sepsis, Peritonitis).
- Thromboembolism (Deep vein thrombosis, Pulmonary embolism).
- Acute Kidney Injury (AKI) & Chronic Kidney Disease (CKD).
- Relapse & Frequent Hospitalization.
Prognosis:
- Minimal Change Disease (MCD): 90% respond well to steroids.
- FSGS & Congenital Nephrotic Syndrome: Poor response, may progress to ESRD.
- Long-term follow-up required to monitor kidney function.
Key Points:
✔ Nephrotic Syndrome is a kidney disorder characterized by proteinuria, edema, hypoalbuminemia, and hyperlipidemia.
✔ Minimal Change Disease (MCD) is the most common type in children and responds well to steroids.
✔ Treatment includes corticosteroids, diuretics, antihypertensives, and immunosuppressants in resistant cases.
✔ Nursing care focuses on fluid monitoring, infection prevention, and medication adherence.
✔ Early diagnosis and proper treatment improve outcomes, but relapses may occur.
Acute Glomerulonephritis in Children
Definition:
Acute Glomerulonephritis (AGN) is a sudden onset of inflammation of the glomeruli (the tiny filters in the kidneys), leading to proteinuria, hematuria, hypertension, and reduced kidney function. It is commonly seen after a streptococcal infection in children.
Etiology (Causes & Risk Factors):
1. Infectious Causes (Post-Infectious Glomerulonephritis)
- Post-Streptococcal Glomerulonephritis (PSGN) – Most common in children, occurs 1-2 weeks after a throat infection (Strep Throat) or 3-6 weeks after a skin infection (Impetigo).
- Bacterial Infections (Staphylococcus, Pneumococcus).
- Viral Infections (Hepatitis B, C, HIV, Epstein-Barr Virus).
- Parasitic Infections (Malaria, Schistosomiasis).
2. Autoimmune & Immune-Mediated Causes
- IgA Nephropathy (Berger’s Disease).
- Systemic Lupus Erythematosus (SLE).
- Henoch-Schönlein Purpura (HSP).
3. Other Causes
- Vasculitis (Wegener’s Granulomatosis).
- Membranoproliferative Glomerulonephritis (MPGN).
4. Risk Factors:
- Recent streptococcal throat or skin infection.
- Family history of kidney disease.
- Children between ages 5-12 years.
- Poor hygiene and overcrowded living conditions.
Types of Acute Glomerulonephritis:
| Type | Cause | Key Features |
|---|---|---|
| Post-Streptococcal Glomerulonephritis (PSGN) | Group A Streptococcus infection | Hematuria, Edema, Hypertension |
| IgA Nephropathy (Berger’s Disease) | IgA deposition in glomeruli | Recurrent gross hematuria, Usually follows an upper respiratory infection |
| Lupus Nephritis | Systemic Lupus Erythematosus (SLE) | Autoimmune disorder, Severe proteinuria |
| Henoch-Schönlein Purpura (HSP) | Small-vessel vasculitis | Purpuric rash, Joint pain, Abdominal pain, Hematuria |
Pathophysiology:
- Immune Complex Deposition:
- Antigen-antibody complexes (Post-infection or autoimmune) deposit in the glomeruli, triggering an immune response.
- Inflammation of the Glomerular Membrane:
- Causes capillary damage, swelling, and thickening of the filtration barrier.
- Reduced Glomerular Filtration Rate (GFR):
- Leads to fluid retention, hypertension, and oliguria.
- Protein & Blood Leakage into Urine:
- Results in proteinuria and hematuria (cola-colored urine).
- Sodium & Water Retention:
- Causes generalized edema, particularly in the face and lower limbs.
Clinical Manifestations (Signs & Symptoms):
1. Classic Symptoms (PSGN Triad)
- Hematuria (“Cola-colored” or “Tea-colored” urine).
- Hypertension (High Blood Pressure).
- Edema (Swelling of the face, periorbital region, legs).
2. Additional Symptoms:
- Oliguria (Decreased urine output).
- Mild to Moderate Proteinuria.
- Headache & Irritability (Due to hypertension).
- Fatigue & Malaise.
- Fever (If associated with infection).
3. Severe Cases:
- Severe Hypertension → Risk of Seizures (Hypertensive Encephalopathy).
- Pulmonary Edema → Shortness of breath, Crackles in lungs.
- Heart Failure due to fluid overload.
Diagnosis:
1. Laboratory Tests:
- Urinalysis:
- Hematuria (Red Blood Cell Casts).
- Mild to moderate proteinuria.
- Blood Tests:
- Increased Blood Urea Nitrogen (BUN) & Creatinine (Indicates reduced kidney function).
- Low Complement Levels (C3) (In PSGN).
- Positive Anti-Streptolysin O (ASO) Titers (For Post-Streptococcal Glomerulonephritis).
- Electrolytes:
- Hyperkalemia, Hyponatremia due to kidney dysfunction.
2. Imaging Studies:
- Renal Ultrasound:
- Shows swollen kidneys in acute cases.
3. Kidney Biopsy (For Severe or Atypical Cases):
- Indicated when nephrotic symptoms, rapid progression, or persistent kidney dysfunction occur.
Medical Management (Non-Surgical Treatment):
1. Supportive Therapy (Mild Cases)
- Bed Rest Until Edema & Hypertension Improve.
- Fluid Restriction (For Oliguria & Severe Edema).
- Low-Sodium & Low-Potassium Diet.
2. Medications:
- Diuretics (Furosemide):
- For fluid overload & hypertension.
- Antihypertensive Drugs (ACE Inhibitors, Calcium Channel Blockers):
- To control high blood pressure.
- Corticosteroids (Prednisolone):
- Used in IgA Nephropathy, Lupus Nephritis.
- Antibiotics (Penicillin, Amoxicillin):
- Only if active streptococcal infection is present (Not useful after PSGN develops).
3. Dialysis (For Severe Cases)
- Indications:
- Severe renal failure (Creatinine >5 mg/dL).
- Uncontrolled fluid overload (Pulmonary edema).
- Hyperkalemia (Life-threatening potassium levels).
Surgical Management:
- Not required in most cases.
- Kidney Transplant may be necessary in cases of rapid progression to End-Stage Renal Disease (ESRD).
Nursing Management:
1. Pre-Treatment Nursing Care:
- Monitor fluid status (Daily weight, Intake-output measurement).
- Assess for signs of severe hypertension (Headache, Blurred vision, Seizures).
- Encourage rest to reduce metabolic demand on kidneys.
2. Nursing Care During Treatment:
A. Medication Administration
- Give diuretics and antihypertensives as prescribed.
- Monitor for side effects (Hypotension, Electrolyte imbalances).
B. Monitoring & Early Complication Prevention
- Monitor urine output (Signs of worsening kidney function).
- Watch for fluid overload symptoms (Lung crackles, Shortness of breath).
C. Dietary Management
- Low-sodium, low-potassium diet.
- Encourage small, frequent meals for appetite maintenance.
D. Infection Prevention
- Promote hand hygiene.
- Encourage vaccinations (Pneumococcal, Influenza).
Complications of Acute Glomerulonephritis:
- Hypertensive Encephalopathy (Seizures, Confusion).
- Pulmonary Edema (Fluid in lungs).
- Acute Kidney Injury (AKI) → Chronic Kidney Disease (CKD).
- Heart Failure (Due to fluid overload).
Prognosis:
- Post-Streptococcal Glomerulonephritis (PSGN) has a Good Prognosis (95% Recovery).
- IgA Nephropathy & Lupus Nephritis may progress to Chronic Kidney Disease.
- Early diagnosis & blood pressure control improve long-term outcomes.
Key Points:
✔ Acute Glomerulonephritis is commonly post-infectious and presents with hematuria, hypertension, and edema.
✔ Diagnosis includes urinalysis, ASO titers, and kidney function tests.
✔ Treatment focuses on blood pressure control, diuretics, fluid restriction, and infection prevention.
✔ Nursing care includes monitoring urine output, edema, and preventing complications.
Renal Failure in Children
Definition:
Renal failure in children is a condition where the kidneys lose their ability to filter waste, regulate fluid balance, and maintain electrolyte homeostasis. It can be acute (sudden onset) or chronic (progressive loss of function), leading to metabolic imbalances, fluid overload, and uremia.
Etiology (Causes & Risk Factors):
1. Acute Kidney Injury (AKI) – Sudden, Reversible Kidney Failure
Causes:
- Pre-Renal (Most Common in Children – Due to Decreased Blood Flow)
- Severe dehydration (Vomiting, Diarrhea, Hypovolemia).
- Sepsis (Severe infection leading to shock).
- Congenital heart disease (Reduced kidney perfusion).
- Intrinsic Renal (Due to Direct Kidney Damage)
- Acute Glomerulonephritis (Post-Streptococcal).
- Hemolytic Uremic Syndrome (HUS) – Caused by E. coli infections.
- Nephrotoxic drugs (NSAIDs, Aminoglycosides, Chemotherapy).
- Post-Renal (Due to Urinary Tract Obstruction)
- Posterior urethral valves (PUV) – Common in newborn boys.
- Kidney stones, Tumors compressing the ureters.
2. Chronic Kidney Disease (CKD) – Progressive, Irreversible Kidney Damage
Causes:
- Congenital Anomalies (Most Common Cause in Children)
- Renal Dysplasia, Reflux Nephropathy.
- Glomerular Diseases
- Focal Segmental Glomerulosclerosis (FSGS), IgA Nephropathy.
- Systemic Diseases
- Lupus Nephritis, Diabetes Mellitus.
- Inherited Conditions
- Polycystic Kidney Disease (PKD), Alport Syndrome.
Types of Renal Failure:
| Type | Cause | Onset | Reversibility |
|---|---|---|---|
| Acute Kidney Injury (AKI) | Sudden dehydration, infections, toxins | Hours to Days | Reversible if treated early |
| Chronic Kidney Disease (CKD) | Congenital, genetic, chronic infections | Months to Years | Irreversible, progresses to ESRD |
| End-Stage Renal Disease (ESRD) | Complete kidney failure | Permanent | Requires dialysis or transplant |
Pathophysiology:
1. Acute Kidney Injury (AKI)
- Pre-Renal Failure:
- Decreased blood flow to the kidneys → Ischemia → Reduced urine output (Oliguria).
- Intrinsic Renal Failure:
- Inflammation, toxins, or infection cause kidney tissue damage → Proteinuria, Hematuria.
- Post-Renal Failure:
- Urine backs up due to obstruction → Hydronephrosis → Kidney damage.
2. Chronic Kidney Disease (CKD)
- Slow loss of nephrons over time → Progressive decline in glomerular filtration rate (GFR).
- Retention of waste products → Uremia (High blood urea and creatinine).
- Electrolyte imbalances → Hyperkalemia, Metabolic Acidosis.
- Loss of erythropoietin → Anemia.
- Bone disorders (Renal Osteodystrophy) due to impaired vitamin D metabolism.
Clinical Manifestations (Signs & Symptoms):
1. Acute Kidney Injury (AKI) Symptoms
- Oliguria (Decreased urine output, <1mL/kg/hr).
- Fluid overload (Edema, Hypertension).
- Hyperkalemia (Irregular heartbeat, Weakness).
- Nausea, Vomiting, Fatigue.
2. Chronic Kidney Disease (CKD) Symptoms
- Polyuria (Excessive urination in early stages).
- Growth retardation, Failure to thrive (Delayed development).
- Anemia (Pale skin, Fatigue, Shortness of breath).
- Bone pain (Due to calcium and phosphate imbalance).
- Uremic Symptoms (Itchy skin, Bad breath, Loss of appetite).
Diagnosis:
1. Laboratory Tests:
- Blood Urea Nitrogen (BUN) & Creatinine: Elevated in renal failure.
- Serum Electrolytes: Hyperkalemia, Hyponatremia, Metabolic Acidosis.
- Complete Blood Count (CBC): Anemia (Low Hemoglobin & RBCs).
- Urinalysis: Proteinuria, Hematuria, Casts.
2. Imaging Studies:
- Renal Ultrasound: Identifies kidney size, hydronephrosis, or congenital anomalies.
- Voiding Cystourethrogram (VCUG): Evaluates vesicoureteral reflux.
- MRI/CT Scan: Detailed assessment of kidney structure.
3. Renal Biopsy:
- For chronic kidney disease or suspected glomerulonephritis.
Medical Management (Non-Surgical Treatment):
1. Supportive Treatment (For AKI & CKD)
- Fluid Restriction (If Oliguric).
- Sodium & Potassium Restriction (To prevent hyperkalemia).
- Calcium & Vitamin D Supplements (For Bone Health).
- Iron & Erythropoietin Therapy (For Anemia).
2. Medications:
- Diuretics (Furosemide): To remove excess fluid.
- Antihypertensives (ACE Inhibitors, Beta-Blockers): To control blood pressure.
- Sodium Bicarbonate: To correct metabolic acidosis.
3. Dialysis (For Severe Kidney Failure)
- Peritoneal Dialysis (PD) (Preferred in children).
- Hemodialysis (HD) (For ESRD patients).
Surgical Management:
- Vesicostomy/Ureterostomy: For obstructive uropathy.
- Kidney Transplant: Definitive treatment for ESRD.
Nursing Management:
1. Pre-Diagnosis Nursing Care:
- Monitor urine output (Oliguria vs. Polyuria).
- Assess for edema, respiratory distress (Pulmonary edema).
- Measure blood pressure regularly.
2. During Treatment:
A. Medication Administration
- Administer diuretics, antihypertensives, and erythropoietin as prescribed.
- Monitor for medication side effects (Hyperkalemia, Hypotension).
B. Fluid & Electrolyte Management
- Monitor for signs of fluid overload (Swelling, Lung crackles).
- Maintain strict input-output balance.
C. Nutritional Support
- Encourage a low-sodium, low-potassium diet.
- Provide high-calorie, low-protein meals in CKD.
D. Dialysis Care
- Ensure proper peritoneal dialysis catheter care.
- Monitor for infection (Peritonitis, Fever, Abdominal pain).
3. Psychosocial & Family Support
- Educate parents about renal disease management.
- Prepare the child and family for long-term dialysis or kidney transplant.
- Provide emotional support for growth and development concerns.
Complications of Renal Failure:
- Severe Hypertension → Stroke, Heart Failure.
- Electrolyte Imbalances → Hyperkalemia (Life-threatening arrhythmias).
- Bone Disease → Osteoporosis, Fractures.
- Infections → Due to suppressed immunity.
- Growth Retardation → Poor physical development in children.
Prognosis:
- AKI is reversible if treated early.
- CKD progresses to ESRD, requiring lifelong dialysis or kidney transplant.
- Early management improves quality of life.
Key Points:
✔ Renal failure in children can be acute (AKI) or chronic (CKD).
✔ Early diagnosis and supportive care improve prognosis.
✔ Dialysis or kidney transplant is required in End-Stage Renal Disease (ESRD).
✔ Nursing care includes fluid monitoring, infection prevention, and dietary management.
Spina Bifida in Children –
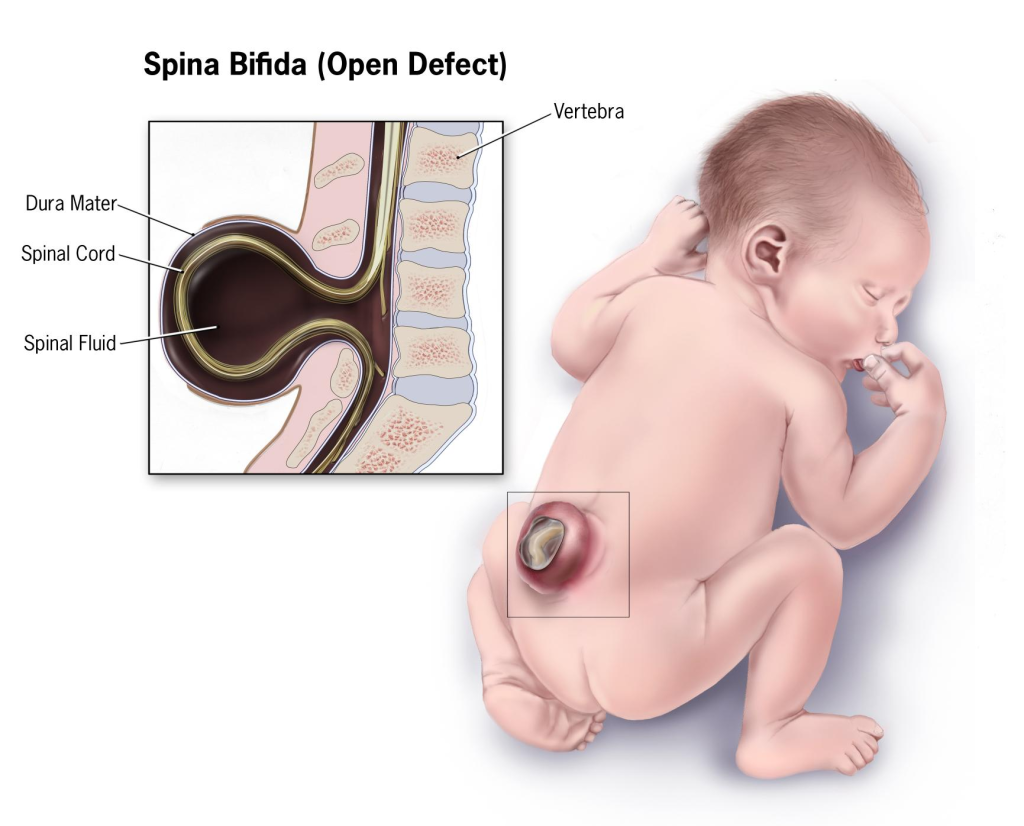
Definition:
Spina bifida is a congenital neural tube defect (NTD) in which the vertebrae fail to close completely around the spinal cord, leading to varying degrees of nerve damage and disability. It occurs during the first 3–4 weeks of fetal development when the neural tube fails to close properly. This defect can cause motor and sensory impairments, bladder and bowel dysfunction, and orthopedic complications.
Etiology (Causes & Risk Factors):
1. Genetic Factors:
- Family history of neural tube defects (NTDs).
- Gene mutations affecting folate metabolism.
2. Environmental & Nutritional Factors:
- Folic Acid Deficiency (Most common preventable cause).
- Folic acid is crucial for DNA synthesis and neural tube closure.
- Women who do not consume enough folic acid before conception have a higher risk of having a baby with spina bifida.
- Maternal Diabetes & Obesity:
- Poorly controlled diabetes increases the risk of neural tube defects.
- Maternal Hyperthermia (Fever or Sauna Use in Early Pregnancy):
- High temperatures may disrupt neural tube closure.
- Maternal Use of Certain Medications During Pregnancy:
- Anticonvulsants (Valproic acid, Carbamazepine).
- Methotrexate (Folate antagonist used in cancer treatment).
- Exposure to Toxins & Radiation During Pregnancy:
- Chemicals and radiation may interfere with neural tube development.
3. Risk Factors:
- Maternal age (Teenage pregnancies and women over 35 have a higher risk).
- Low socioeconomic status (Poor maternal nutrition).
- Ethnicity (Hispanic and Caucasian women have a slightly higher risk).
Types of Spina Bifida:
| Type | Severity | Description |
|---|---|---|
| Spina Bifida Occulta | Mildest form | Small defect in vertebrae, No visible sac, Often asymptomatic |
| Meningocele | Moderate | Meninges protrude through spinal opening, but spinal cord remains intact, Low risk of nerve damage |
| Myelomeningocele (Open Spina Bifida) | Severe | Spinal cord and meninges protrude, causing nerve damage, paralysis, bowel & bladder dysfunction |
Pathophysiology:
- Neural Tube Closure Failure (During 3rd-4th week of gestation).
- The neural tube fails to close completely, leaving part of the spinal cord exposed or improperly enclosed in the vertebrae.
- Vertebral Defect Formation:
- The vertebrae fail to develop properly around the spinal cord, forming a bony defect.
- Exposure of Spinal Cord & Nerve Damage:
- In meningocele and myelomeningocele, the spinal cord or meninges protrude through the defect, leading to nerve dysfunction.
- Cerebrospinal Fluid (CSF) Imbalance & Hydrocephalus:
- Abnormal CSF drainage leads to hydrocephalus, requiring shunt placement.
- Bladder & Bowel Dysfunction:
- Due to disrupted nerve supply to the bladder and rectum, leading to urinary incontinence and constipation.
Clinical Manifestations (Signs & Symptoms):
1. Spina Bifida Occulta (Mildest Form)
- No visible sac or protrusion.
- Small dimple, birthmark, or tuft of hair over the lower back.
- Usually asymptomatic but may cause mild back pain or weakness.
2. Meningocele
- Visible fluid-filled sac protruding from the back.
- No spinal cord involvement → Usually no paralysis.
- Mild neurological symptoms, if any.
3. Myelomeningocele (Most Severe Form)
- Large sac containing spinal cord & nerves protruding from the back.
- Partial or complete paralysis below the defect.
- Loss of bladder & bowel control (Neurogenic bladder and bowel).
- Hydrocephalus (Increased CSF in the brain → Requires VP Shunt).
- Musculoskeletal abnormalities (Clubfoot, Hip dislocation, Scoliosis).
- Chiari II Malformation (Part of the brainstem and cerebellum herniate into the spinal canal, affecting breathing and swallowing).
Diagnosis:
1. Prenatal Diagnosis (Before Birth)
- Maternal Serum Alpha-Fetoprotein (MSAFP) Test:
- Elevated levels suggest neural tube defects.
- Ultrasound (16–18 weeks of gestation):
- Detects open neural tube defects, hydrocephalus, and spinal abnormalities.
- Amniocentesis:
- Measures Alpha-Fetoprotein (AFP) and Acetylcholinesterase (AChE) in amniotic fluid.
2. Postnatal Diagnosis (After Birth)
- Physical Examination:
- Presence of sac-like protrusion, motor deficits, and bladder/bowel dysfunction.
- MRI/CT Scan of the Spine:
- Determines extent of spinal cord involvement.
- Renal Ultrasound & Voiding Cystourethrogram (VCUG):
- Evaluate urinary function and kidney health.
Medical Management (Non-Surgical Treatment):
1. Prenatal Folic Acid Supplementation (Prevention)
- Women planning pregnancy: 400 mcg/day.
- During pregnancy (First trimester): 600 mcg/day.
- High-risk women (Previous NTD pregnancy): 4 mg/day.
2. Hydrocephalus Management
- Ventriculoperitoneal (VP) Shunt (To drain excess CSF).
3. Bladder & Bowel Management
- Intermittent catheterization for bladder emptying.
- Stool softeners & High-fiber diet for bowel regulation.
4. Physiotherapy & Mobility Support
- Braces, Walkers, or Wheelchairs for mobility.
- Physical therapy to improve muscle strength and prevent contractures.
Surgical Management:
1. Prenatal Surgery (Fetal Repair)
- Performed between 19-25 weeks of gestation.
- Reduces risk of hydrocephalus and improves leg function.
2. Postnatal Surgery (Neonatal Repair)
- Performed within 24-48 hours after birth.
- Closes the spinal defect to prevent infection and further nerve damage.
3. Hydrocephalus Treatment
- VP Shunt Placement (To manage cerebrospinal fluid imbalance).
4. Orthopedic Surgeries
- Corrects clubfoot, scoliosis, or hip dislocation.
Nursing Management:
1. Preoperative Nursing Care:
- Keep the exposed sac covered with a sterile, moist saline dressing.
- Position the infant in prone (on the stomach) to prevent pressure on the sac.
- Monitor for signs of infection (Fever, Redness, CSF leakage).
2. Postoperative Nursing Care:
- Monitor for signs of hydrocephalus (Bulging fontanelles, Vomiting).
- Assess for CSF leakage from the surgical site.
- Encourage passive range of motion (ROM) exercises for leg movement.
3. Long-Term Nursing Care:
- Teach parents catheterization techniques for bladder management.
- Encourage high-fiber diets & bowel training programs.
- Monitor for signs of shunt malfunction (Headache, Vomiting, Seizures).
Prognosis:
- Mild cases (Spina Bifida Occulta) have normal life expectancy.
- Myelomeningocele cases require lifelong care (Mobility aids, Shunt management).
- Prenatal surgery improves outcomes, but challenges remain.
Hydrocephalus in Children
Definition:
Hydrocephalus is a neurological condition characterized by excessive accumulation of cerebrospinal fluid (CSF) within the ventricles of the brain, leading to increased intracranial pressure (ICP) and enlargement of the head in infants. It occurs due to an imbalance in CSF production, flow, or absorption.
Etiology (Causes & Risk Factors):
1. Congenital Causes (Present at Birth)
- Neural Tube Defects (Spina Bifida, Myelomeningocele).
- Aqueductal Stenosis (Narrowing of the cerebral aqueduct).
- Arnold-Chiari Malformation (Herniation of brain tissue into the spinal canal).
- Dandy-Walker Syndrome (Cyst formation in the cerebellum).
- Genetic Mutations affecting brain development.
2. Acquired Causes (After Birth)
- Infections (Meningitis, Encephalitis).
- Brain Hemorrhage (Intraventricular hemorrhage in preterm infants).
- Head Trauma (Brain injury leading to obstruction of CSF flow).
- Brain Tumors (Compressing the ventricular system).
3. Risk Factors:
- Premature birth (Increased risk of intraventricular hemorrhage).
- Maternal infections during pregnancy (TORCH infections – Toxoplasmosis, Rubella, Cytomegalovirus, Herpes).
- Family history of congenital hydrocephalus.
Types of Hydrocephalus:
| Type | Cause | Description |
|---|---|---|
| Communicating Hydrocephalus | Impaired CSF absorption | CSF flows freely but is not absorbed properly |
| Non-Communicating (Obstructive) Hydrocephalus | Blocked CSF flow | CSF is obstructed in the ventricular system |
| Congenital Hydrocephalus | Present at birth | Due to genetic defects or developmental anomalies |
| Acquired Hydrocephalus | Develops later | Due to infections, trauma, or tumors |
| Normal Pressure Hydrocephalus (NPH) | Seen in older children | Ventricles enlarge, but ICP remains normal |
Pathophysiology:
- Overproduction of CSF (Rare Cause):
- Increased production by choroid plexus overwhelms absorption capacity.
- Obstruction to CSF Flow (Most Common Cause):
- Aqueductal Stenosis or Tumors block ventricular pathways.
- Leads to enlargement of the ventricles and compression of brain tissue.
- Impaired CSF Absorption:
- Dysfunction of arachnoid villi (Infections, Hemorrhage).
- Increased Intracranial Pressure (ICP):
- Leads to brain compression, impaired circulation, and neurological symptoms.
Clinical Manifestations (Signs & Symptoms):
1. In Infants (Before Skull Sutures Close)
- Bulging fontanelles (Soft spot on head is tense and full).
- Rapidly increasing head circumference (Macrocephaly).
- Prominent scalp veins.
- Setting sun sign (Eyes appear pushed downward, sclera visible above iris).
- High-pitched cry, Irritability.
- Seizures.
2. In Older Children (After Skull Sutures Close)
- Headache (Worsens in the morning due to CSF accumulation overnight).
- Vomiting (Without nausea, projectile in nature).
- Blurred or double vision.
- Difficulty walking, Poor coordination.
- Memory loss, Confusion.
3. Signs of Increased Intracranial Pressure (ICP)
- Bradycardia, Hypertension, and Irregular Breathing (Cushing’s Triad – Late Sign).
- Papilledema (Swelling of the optic disc leading to vision loss).
Diagnosis:
1. Physical Examination:
- Measurement of head circumference (Rapid growth indicates hydrocephalus).
- Neurological assessment (Reflexes, Motor function, Cranial nerve involvement).
2. Imaging Studies:
- Cranial Ultrasound (For newborns and preterm infants).
- CT Scan of the Brain (Detects enlarged ventricles, obstruction).
- MRI of the Brain (More detailed assessment of CSF flow and brain structures).
3. Lumbar Puncture (LP):
- Not done in cases of increased ICP (Risk of brain herniation).
Medical Management (Non-Surgical Treatment):
1. Medications (Temporary Measures)
- Acetazolamide (Carbonic Anhydrase Inhibitor): Reduces CSF production.
- Furosemide (Diuretic): Lowers CSF volume temporarily.
- Corticosteroids: Reduces inflammation in post-infectious hydrocephalus.
2. Supportive Therapy
- Head elevation to reduce ICP.
- Seizure management (Anticonvulsants if needed).
- Nutritional support (Small, frequent feedings in infants).
3. Management of Hydrocephalus in Premature Infants
- Serial lumbar punctures or ventriculostomy (Temporary relief before surgery).
Surgical Management:
1. Ventriculoperitoneal (VP) Shunt (Most Common Surgery)
- Redirects CSF from ventricles to the peritoneal cavity, where it is absorbed.
- Components:
- Ventricular catheter (Placed in lateral ventricle).
- Pressure valve (Regulates CSF flow).
- Peritoneal catheter (Drains CSF into the abdomen).
2. Endoscopic Third Ventriculostomy (ETV)
- Alternative to VP shunt (For obstructive hydrocephalus).
- Creates a hole in the third ventricle to allow CSF drainage.
3. Ventriculoatrial (VA) or Ventriculopleural Shunt
- Used if VP shunt is not feasible (Drains CSF into the heart or pleural cavity).
4. Choroid Plexus Cauterization
- Reduces CSF production in cases of overproduction.
Nursing Management:
1. Preoperative Nursing Care:
- Monitor head circumference daily.
- Assess for signs of increased ICP (Bulging fontanelle, Irritability, Vomiting).
- Position infant with head elevated (30 degrees).
- Ensure hydration & nutrition (Frequent small feedings).
- Prevent infection (Hand hygiene, Antibiotics if needed).
2. Postoperative Nursing Care (After VP Shunt or ETV Surgery):
- Monitor for shunt malfunction (Signs of increased ICP).
- Assess for shunt infection (Fever, Redness, Swelling along shunt tract).
- Keep the infant in a flat position initially, then gradually elevate the head.
- Monitor for CSF leakage from the incision site.
- Assess for abdominal distension (For VP shunt-related peritonitis).
3. Long-Term Nursing Care & Parental Education:
- Teach parents to recognize signs of shunt malfunction (Headache, Vomiting, Lethargy).
- Regular follow-up visits for shunt function monitoring.
- Encourage normal activities while avoiding head trauma.
- Vaccination is essential to prevent infections.
Complications of Hydrocephalus:
- Shunt Malfunction (Obstruction, Disconnection, Over-drainage).
- Shunt Infection (Peritonitis, Meningitis).
- Cognitive Impairment & Learning Disabilities.
- Vision Problems (Optic nerve damage due to ICP).
- Seizures.
Prognosis:
- Early diagnosis & treatment improve outcomes.
- VP shunts or ETV provide long-term symptom control.
- Some children may have mild developmental delays.
Key Points:
✔ Hydrocephalus is caused by an imbalance in CSF production, flow, or absorption.
✔ Congenital causes include neural tube defects, aqueductal stenosis, and Chiari malformation.
✔ Diagnosis includes ultrasound, CT, MRI, and neurological assessment.
✔ Treatment involves VP shunt placement, ETV, or temporary medical management.
✔ Nursing care focuses on monitoring ICP, preventing infections, and parental education.
Meningitis in Children
Definition:
Meningitis is an inflammation of the meninges (protective membranes covering the brain and spinal cord), caused by bacterial, viral, fungal, or parasitic infections. It leads to neurological complications, increased intracranial pressure (ICP), and can be life-threatening if not treated promptly.
Etiology (Causes & Risk Factors):
1. Infectious Causes
A. Bacterial Meningitis (Severe & Life-Threatening)
- Neonates (<1 month):
- Group B Streptococcus (GBS) – Most common.
- Escherichia coli (E. coli).
- Listeria monocytogenes.
- Children (>1 month):
- Streptococcus pneumoniae (Pneumococcus).
- Neisseria meningitidis (Meningococcus) – Highly contagious.
- Haemophilus influenzae type B (Hib) – Rare due to vaccination.
B. Viral (Aseptic) Meningitis (Milder & More Common)
- Enteroviruses (Most Common) – Coxsackievirus, Echovirus.
- Herpes Simplex Virus (HSV-2), Varicella-Zoster Virus.
- Mumps Virus, Measles Virus (If unvaccinated).
C. Fungal Meningitis
- Cryptococcus neoformans – Seen in immunocompromised children (HIV, cancer).
D. Parasitic & Non-Infectious Causes
- Amoebic Meningitis (Naegleria fowleri – Found in contaminated water).
- Tuberculous Meningitis (Mycobacterium tuberculosis).
- Autoimmune Diseases, Drug Reactions.
Risk Factors:
- Premature birth, Low birth weight.
- Lack of vaccination (Hib, Pneumococcal, Meningococcal vaccines).
- Close contact with an infected person (Crowded places, Daycare centers).
- Head trauma, Neurosurgical procedures (CSF leaks).
- Immunodeficiency (HIV, Chemotherapy, Malnutrition).
Types of Meningitis:
| Type | Cause | Severity |
|---|---|---|
| Bacterial Meningitis | Streptococcus pneumoniae, Neisseria meningitidis, Haemophilus influenzae | Severe, life-threatening, requires urgent treatment |
| Viral Meningitis (Aseptic) | Enteroviruses, Herpesvirus, Mumps, Measles | Mild, self-limiting, supportive care needed |
| Fungal Meningitis | Cryptococcus, Candida | Seen in immunocompromised children |
| Tuberculous Meningitis | Mycobacterium tuberculosis | Chronic, Gradual onset, Requires long-term antibiotics |
Pathophysiology:
- Entry of Pathogens into CNS:
- Bacteria/Viruses invade via bloodstream, direct injury (head trauma), or ear/nose infections (Otitis media, Sinusitis).
- Inflammatory Response:
- Immune cells release cytokines (IL-1, TNF-alpha), increasing permeability of the blood-brain barrier (BBB).
- Increased Intracranial Pressure (ICP):
- Brain swelling (Edema) + Blocked CSF flow = Hydrocephalus.
- Neuronal Damage & Complications:
- Seizures, Brain infarctions, Hearing loss, Cognitive deficits.
Clinical Manifestations (Signs & Symptoms):
1. Symptoms in Neonates & Infants (<1 year)
- High fever, Lethargy, Poor feeding, Weak cry.
- Bulging fontanelle (Late sign).
- Seizures, Hypotonia (Weak muscle tone).
- Irritability, High-pitched cry.
2. Symptoms in Older Infants & Children (>1 year)
- Sudden high fever, Severe headache, Vomiting.
- Neck stiffness (Nuchal rigidity).
- Photophobia (Sensitivity to light).
- Seizures, Confusion, Altered mental status.
- Skin rash (Purpuric rash in Meningococcal Meningitis).
3. Signs of Increased Intracranial Pressure (ICP)
- Cushing’s Triad (Bradycardia, Hypertension, Irregular breathing).
- Papilledema (Swelling of the optic nerve, Blurred vision).
Diagnosis:
1. Laboratory Tests:
- Complete Blood Count (CBC): Elevated WBCs.
- Blood Culture: Identifies bacterial infections.
2. Lumbar Puncture (LP) – Gold Standard Test
- Cerebrospinal Fluid (CSF) Analysis: Parameter Bacterial Meningitis Viral Meningitis CSF WBC Count High (>1000 cells/µL) Mildly Elevated (10-1000 cells/µL) CSF Protein High (>100 mg/dL) Normal or slightly elevated CSF Glucose Low (<40 mg/dL) Normal
- Gram Stain & Culture (For bacterial identification).
- PCR (For viral infections like HSV, Enteroviruses).
3. Imaging (If Signs of Increased ICP)
- CT or MRI Brain:
- Detects hydrocephalus, brain abscess, infarcts.
Medical Management (Non-Surgical Treatment):
1. Empirical Antibiotic Therapy (For Bacterial Meningitis)
- Neonates (<1 month):
- Ampicillin + Gentamicin OR Cefotaxime.
- Children (>1 month):
- Ceftriaxone + Vancomycin.
- Dexamethasone (Steroid) for Pneumococcal Meningitis.
2. Antiviral Therapy (For Viral Meningitis)
- Acyclovir (For HSV Meningitis).
- Supportive care (IV fluids, Fever control).
3. Supportive Treatment
- Antipyretics (Paracetamol) for fever.
- Seizure control (Diazepam, Phenytoin).
- IV fluids, Oxygen support if needed.
Surgical Management (For Complications)
- Ventriculoperitoneal (VP) Shunt: For hydrocephalus.
- Craniotomy: For brain abscess drainage.
Nursing Management:
1. Pre-Treatment Nursing Care:
- Initiate droplet isolation (Until bacterial cause is ruled out).
- Monitor vital signs (BP, Temperature, Neurological status).
- Assess for signs of increased ICP (Headache, Bulging fontanelles, Seizures).
- Maintain hydration & Monitor fluid balance.
2. Nursing Care During Treatment:
A. Medication Administration
- Ensure timely administration of antibiotics/antivirals.
- Administer anticonvulsants if seizures occur.
B. Comfort Measures
- Keep the room dim (Reduce photophobia & headache).
- Elevate head 30 degrees (Reduce ICP).
C. Infection Prevention
- Strict hand hygiene, Isolate the child until confirmed non-contagious.
- Encourage vaccination (Hib, Pneumococcal, Meningococcal vaccines).
3. Long-Term Nursing Care & Parental Education:
- Monitor for long-term complications (Hearing loss, Cognitive impairment).
- Encourage follow-up neurological assessments.
Complications of Meningitis:
- Seizures & Coma (Severe cases).
- Hydrocephalus → Requires VP Shunt.
- Hearing Loss (Sensorineural deafness).
- Septic Shock (Meningococcemia).
- Cerebral Edema & Herniation.
Prognosis:
- Bacterial Meningitis → 10-20% mortality if untreated.
- Viral Meningitis → Good prognosis, self-limiting.
Key Points:
✔ Meningitis is a life-threatening condition requiring urgent treatment.
✔ Diagnosis includes lumbar puncture, CSF analysis, and blood culture.
✔ Bacterial meningitis requires IV antibiotics, while viral meningitis is self-limiting.
✔ Nursing care includes infection control, seizure management, and ICP monitoring.
Encephalitis in Children
Definition:
Encephalitis is a serious inflammation of the brain tissue, primarily caused by viral infections, but can also result from bacterial, fungal, parasitic, or autoimmune disorders. It leads to brain swelling, neurological dysfunction, and, in severe cases, long-term complications like seizures and cognitive impairment.
Etiology (Causes & Risk Factors):
1. Infectious Causes
A. Viral Encephalitis (Most Common Cause)
- Herpes Simplex Virus (HSV-1 & HSV-2) – Most common and severe.
- Enteroviruses (Coxsackievirus, Echovirus).
- Arboviruses (West Nile Virus, Japanese Encephalitis Virus, Zika Virus, Dengue Virus).
- Mumps, Measles, Varicella-Zoster Virus (If unvaccinated).
- Rabies Virus (Transmitted through animal bites).
B. Bacterial Encephalitis
- Mycoplasma pneumoniae (Atypical bacterial cause).
- Tuberculous (TB) Encephalitis – Chronic infection due to Mycobacterium tuberculosis.
- Neuroborreliosis (Lyme Disease) – From Borrelia burgdorferi (tick-borne).
C. Fungal & Parasitic Encephalitis
- Cryptococcus, Candida (Seen in immunocompromised children – HIV/AIDS, chemotherapy).
- Toxoplasma gondii (Congenital toxoplasmosis in newborns).
2. Non-Infectious (Autoimmune & Post-Infectious Causes)
- Autoimmune Encephalitis (Anti-NMDA Receptor Encephalitis).
- Post-Infectious Encephalitis (Acute Disseminated Encephalomyelitis – ADEM).
- Vaccine-Related Encephalitis (Rare, seen in live-attenuated vaccines).
Risk Factors:
- Newborns, Infants, & Immunocompromised children.
- Lack of vaccination (Measles, Mumps, Rubella, Japanese Encephalitis).
- Exposure to mosquito-borne or tick-borne viruses.
- Travel to endemic areas (India, Southeast Asia – Japanese Encephalitis).
- Animal exposure (Rabies virus).
Types of Encephalitis:
| Type | Cause | Severity |
|---|---|---|
| Primary (Viral) Encephalitis | Direct viral invasion (HSV, Enterovirus, Arboviruses) | Severe, Can be life-threatening |
| Post-Infectious (Autoimmune) Encephalitis | Immune response to infection (ADEM, Anti-NMDA receptor encephalitis) | Less severe but can cause long-term neurological damage |
| Parasitic/Fungal Encephalitis | Cryptococcus, Toxoplasmosis | Seen in immunocompromised children |
Pathophysiology:
- Viral/Bacterial Entry Into CNS:
- Infection reaches the brain via bloodstream, direct injury, or peripheral nerves.
- Immune-Mediated Inflammatory Response:
- Activation of cytokines (IL-1, TNF-alpha), leading to blood-brain barrier disruption.
- Cerebral edema & neuronal dysfunction occur.
- Increased Intracranial Pressure (ICP):
- Brain swelling, CSF obstruction → Hydrocephalus (In some cases).
- Neuronal Injury & Neurological Symptoms:
- Seizures, Altered consciousness, Cognitive impairment.
Clinical Manifestations (Signs & Symptoms):
1. Early Symptoms (Mild Cases)
- Fever, Headache, Malaise, Fatigue.
- Nausea, Vomiting, Poor appetite.
- Irritability (In infants).
2. Severe Symptoms (Advanced Cases)
- High fever (>39°C).
- Seizures (Generalized or focal).
- Altered consciousness (Confusion, Coma in severe cases).
- Photophobia (Sensitivity to light).
- Neck stiffness (If meningitis coexists).
3. Signs of Increased Intracranial Pressure (ICP)
- Cushing’s Triad (Hypertension, Bradycardia, Irregular breathing).
- Bulging fontanelle (In infants).
- Papilledema (Optic disc swelling, Blurred vision).
Diagnosis:
1. Laboratory Tests:
- Complete Blood Count (CBC): Elevated WBCs (Infections).
- Serum Electrolytes: Hyponatremia (SIADH associated with encephalitis).
- Blood Culture: To identify bacterial causes.
2. Lumbar Puncture (LP) – Gold Standard Test
- Cerebrospinal Fluid (CSF) Analysis: Parameter Viral Encephalitis Bacterial Encephalitis CSF WBC Count Mildly Elevated (10-1000 cells/µL) Very High (>1000 cells/µL) CSF Protein Normal or Slightly High High (>100 mg/dL) CSF Glucose Normal Low (<40 mg/dL)
- PCR for Viral Detection: HSV, Enterovirus, Arboviruses.
3. Imaging (MRI/CT Scan)
- Detects brain inflammation, swelling, hydrocephalus, infarcts.
- Hemorrhagic necrosis in HSV Encephalitis (Temporal lobes affected).
4. EEG (Electroencephalography)
- Identifies seizure activity or abnormal brain function.
Medical Management (Non-Surgical Treatment):
1. Empirical Antiviral Therapy (For Viral Encephalitis)
- Acyclovir (IV) – For HSV Encephalitis (Best if given within 48 hours).
- Ganciclovir/Foscarnet – For CMV Encephalitis.
2. Antibiotic Therapy (If Bacterial Cause Suspected)
- Ceftriaxone + Vancomycin (For Bacterial Meningoencephalitis).
3. Supportive Treatment
- Fever Control (Paracetamol, Cooling blankets).
- Seizure Control (Diazepam, Levetiracetam, Phenytoin).
- IV Fluids & Electrolyte Correction (SIADH management).
- Oxygen Therapy & Ventilation (For severe cases).
Surgical Management (For Complications)
- Ventriculoperitoneal (VP) Shunt: If hydrocephalus develops.
- Craniotomy: For brain abscess drainage (If secondary infection).
Nursing Management:
1. Pre-Treatment Nursing Care:
- Monitor vital signs, Neurological assessment (Glasgow Coma Scale – GCS).
- Administer antiviral/antibiotics promptly.
- Initiate seizure precautions (Padding, Side positioning).
- Maintain head elevation (30°) to reduce ICP.
2. Nursing Care During Treatment:
A. Medication Administration
- Ensure timely administration of antivirals/antibiotics.
- Monitor for side effects (Nephrotoxicity in Acyclovir).
B. Comfort Measures
- Keep the room dim (Reduces photophobia & headache).
- Prevent dehydration & Malnutrition (IV fluids, NG tube feeding if needed).
C. Infection Prevention
- Hand hygiene, Standard precautions.
- Encourage vaccinations (Measles, Mumps, Rubella, Japanese Encephalitis, Rabies).
3. Long-Term Nursing Care & Parental Education:
- Monitor for neurological complications (Seizures, Cognitive delays).
- Encourage follow-up neurodevelopmental assessments.
Complications of Encephalitis:
- Seizures, Coma, Death (Severe cases).
- Cognitive Impairment, Memory Loss.
- Paralysis, Speech Impairments.
- Hydrocephalus → VP Shunt may be needed.
Prognosis:
- Mild cases (Viral Encephalitis) recover well.
- Severe cases (HSV Encephalitis) may lead to permanent brain damage.
Key Points:
✔ Encephalitis is brain inflammation caused by viral, bacterial, or autoimmune disorders.
✔ Diagnosis includes CSF analysis, MRI, PCR tests.
✔ Treatment involves antivirals (Acyclovir), seizure control, and supportive therapy.
✔ Nursing care includes monitoring ICP, preventing complications, and family education.
Convulsive Disorders in Children: Convulsions and Seizures
Definition:
Convulsions and seizures are sudden, uncontrolled electrical disturbances in the brain that cause changes in behavior, movement, sensation, and consciousness. Convulsions specifically refer to involuntary muscle contractions that occur during certain types of seizures.
Etiology (Causes & Risk Factors):
1. Neurological Causes:
- Epilepsy (Chronic seizure disorder).
- Brain injury (Trauma, Birth asphyxia, Stroke).
- Congenital brain malformations.
- Neurodegenerative diseases (Tay-Sachs, Rett Syndrome).
2. Infectious Causes:
- Meningitis, Encephalitis.
- Neurocysticercosis (Parasitic infection).
- Sepsis with CNS involvement.
3. Metabolic & Genetic Causes:
- Hypoglycemia (Low blood sugar).
- Hypocalcemia, Hypomagnesemia, Hyponatremia.
- Pyridoxine (Vitamin B6) deficiency in neonates.
4. Febrile Seizures (Most Common in Children 6 months–5 years)
- Triggered by high fever (>38.5°C or 101.3°F).
- Usually associated with viral infections (Influenza, Roseola).
- Benign, resolves without long-term epilepsy risk (Simple febrile seizures).
5. Other Causes:
- Head trauma (Accidents, Falls).
- Toxic exposure (Lead poisoning, Drug overdose).
- Cerebral hypoxia (Lack of oxygen during birth).
- Genetic epilepsy syndromes (Dravet syndrome, Lennox-Gastaut syndrome).
Types of Seizures:
1. Generalized Seizures (Affect both hemispheres of the brain)
| Type | Description | Clinical Features |
|---|---|---|
| Tonic-Clonic (Grand Mal) Seizures | Most common, involves full-body convulsions | Loss of consciousness, stiffening (tonic) → jerking (clonic) movements |
| Absence (Petit Mal) Seizures | Brief episodes of staring (Common in children) | No convulsions, sudden unresponsiveness, blinking |
| Myoclonic Seizures | Sudden muscle jerks | Short, shock-like jerks in arms/legs |
| Atonic Seizures (Drop Attacks) | Sudden loss of muscle tone | Child suddenly collapses |
| Tonic Seizures | Continuous muscle stiffness | Stiffening of arms/legs, falls without jerking |
2. Focal (Partial) Seizures (Affect only one part of the brain)
| Type | Description | Clinical Features |
|---|---|---|
| Simple Partial Seizures | No loss of consciousness | Unusual sensations, jerking in one limb |
| Complex Partial Seizures | Impaired consciousness | Staring, repetitive movements (Lip-smacking, picking at clothes) |
| Focal to Bilateral Tonic-Clonic | Starts in one hemisphere, spreads to both | Progresses to full-body convulsions |
Pathophysiology:
- Abnormal electrical activity arises from hyperexcitable neurons in the brain.
- Depolarization spreads → Triggers widespread electrical discharge.
- Excitatory neurotransmitters (Glutamate) are released excessively.
- Inhibitory GABAergic control is reduced, leading to continuous neuronal firing.
- Clinical manifestations (Convulsions, Altered awareness, Motor dysfunction) appear.
Clinical Manifestations (Signs & Symptoms):
1. Pre-Seizure (Aura Phase – Only in Focal Seizures)
- Strange sensations (Tingling, Smells, Tastes).
- Visual changes (Flashes of light, Distorted images).
- Unusual emotions (Fear, Déjà vu).
2. Ictal Phase (During Seizure)
- Loss of consciousness (In generalized seizures).
- Tonic phase (Muscle stiffening, Eyes rolling up, Cyanosis).
- Clonic phase (Rhythmic jerking, Frothing at the mouth).
- Autonomic symptoms (Sweating, Increased heart rate).
- Urinary/Bowel incontinence (During tonic-clonic seizures).
3. Postictal Phase (After Seizure)
- Drowsiness, Confusion, Disorientation.
- Headache, Muscle soreness.
- Brief memory loss, Weakness.
Diagnosis:
1. Clinical History & Physical Examination
- Age of onset, frequency, duration, triggers.
- Family history of epilepsy.
- Neurological examination (Reflexes, Muscle tone, Coordination).
2. Electroencephalogram (EEG) – Gold Standard Test
- Detects abnormal electrical activity in the brain.
- Helps differentiate between focal and generalized seizures.
3. Neuroimaging (MRI/CT Scan)
- Identifies structural abnormalities (Tumors, Stroke, Hydrocephalus).
4. Laboratory Tests
- Blood glucose, Electrolytes, Calcium, Magnesium.
- Lumbar puncture (If infection is suspected).
Medical Management (Non-Surgical Treatment):
1. Acute Seizure Management (Emergency Care)
- Ensure airway patency, turn child to the side (Prevent aspiration).
- Administer IV benzodiazepines (Diazepam, Lorazepam) to stop prolonged seizures.
- Monitor oxygen saturation, provide oxygen if needed.
2. Antiepileptic Drugs (AEDs) – Long-Term Management
| Seizure Type | First-Line Medications |
|---|---|
| Generalized Tonic-Clonic | Valproate, Lamotrigine, Levetiracetam |
| Absence Seizures | Ethosuximide, Valproate |
| Myoclonic Seizures | Valproate, Clonazepam |
| Focal Seizures | Carbamazepine, Phenytoin |
3. Dietary Therapy
- Ketogenic Diet (High-fat, low-carb diet) – Used for drug-resistant epilepsy.
4. Lifestyle Modifications
- Avoid seizure triggers (Sleep deprivation, Flashing lights).
- Encourage a regular sleep schedule.
Surgical Management (For Drug-Resistant Epilepsy)
- Focal Resection Surgery (Removal of epileptic focus).
- Corpus Callosotomy (For atonic seizures, prevents seizure spread).
- Vagus Nerve Stimulation (VNS) – Implanted device that reduces seizure frequency.
Nursing Management:
1. During Seizure (Acute Care)
- Stay calm, ensure patient safety.
- Turn the child to the side (Prevent aspiration).
- Do NOT restrain or insert anything into the mouth.
- Time the seizure duration (If >5 min, call emergency services).
2. Post-Seizure Nursing Care
- Assess vital signs, neurological status.
- Reassure the child & parents.
- Document seizure details (Duration, Type, Triggers).
- Ensure child gets adequate rest.
3. Long-Term Nursing Care & Parental Education
- Medication adherence (Take AEDs as prescribed).
- Identify and avoid triggers.
- Teach caregivers about seizure first-aid.
- Ensure school safety plans for seizure management.
Complications of Seizures:
- Status Epilepticus (Seizures lasting >5 minutes – Medical Emergency).
- Cognitive impairment & Learning difficulties.
- Injury from falls (Head trauma, Fractures).
- Sudden Unexpected Death in Epilepsy (SUDEP – Rare but serious).
Prognosis:
- Febrile seizures have an excellent prognosis (No long-term epilepsy risk).
- Well-controlled epilepsy allows children to lead normal lives.
- Severe epilepsy syndromes (Lennox-Gastaut) have a poorer prognosis.
Key Points:
✔ Seizures result from abnormal brain electrical activity, causing convulsions or altered consciousness.
✔ Diagnosis includes EEG, MRI, and metabolic testing.
✔ Treatment includes antiepileptic drugs (AEDs), ketogenic diet, and surgery (If drug-resistant).
✔ Nursing care focuses on seizure safety, medication adherence, and patient education.
Cerebral Palsy in Children
Definition:
Cerebral Palsy (CP) is a non-progressive neurological disorder caused by brain damage or abnormal brain development before, during, or shortly after birth, affecting movement, muscle tone, posture, and coordination. It is the most common motor disability in childhood and can also involve speech, cognition, vision, and hearing impairments.
Etiology (Causes & Risk Factors):
1. Prenatal Causes (Before Birth – Most Common)
- Intrauterine infections (TORCH infections – Toxoplasmosis, Rubella, Cytomegalovirus, Herpes).
- Maternal diabetes, hypertension, or drug use.
- Multiple pregnancies (Twins, Triplets).
- Genetic mutations affecting brain development.
- Hypoxic-Ischemic Encephalopathy (HIE) due to placental insufficiency.
2. Perinatal Causes (During Birth)
- Birth asphyxia (Lack of oxygen during delivery).
- Prematurity (<32 weeks gestation – Higher risk).
- Low birth weight (<2500g).
- Prolonged labor, Umbilical cord prolapse.
- Neonatal stroke.
3. Postnatal Causes (After Birth)
- Severe neonatal jaundice (Kernicterus – High bilirubin levels).
- Brain infections (Meningitis, Encephalitis).
- Head trauma (Shaken baby syndrome, Accidents).
- Severe dehydration leading to metabolic imbalances.
Types of Cerebral Palsy:
| Type | Cause | Clinical Features |
|---|---|---|
| Spastic CP (Most Common – 70-80%) | Damage to the motor cortex | Muscle stiffness (Hypertonia), Contractures, Scissor gait |
| Dyskinetic (Athetoid) CP | Damage to the basal ganglia | Involuntary movements, Poor coordination |
| Ataxic CP | Damage to the cerebellum | Balance & Coordination problems, Intention tremors |
| Mixed CP | Combination of spastic and athetoid CP | Mixed symptoms of stiffness and involuntary movements |
Pathophysiology:
- Brain Injury or Maldevelopment:
- Hypoxia, Ischemia, or Infection affects the developing brain.
- Neuron Damage in Motor Control Areas:
- Motor cortex → Spasticity, Hyperreflexia.
- Basal ganglia → Involuntary movements (Dyskinetic CP).
- Cerebellum → Ataxia, Balance problems.
- Abnormal Muscle Activation:
- Poor coordination, spasticity (Hypertonia), or involuntary movements (Athetosis).
- Secondary Complications Develop Over Time:
- Joint contractures, Scoliosis, Hip dislocation, Difficulty swallowing (Dysphagia).
Clinical Manifestations (Signs & Symptoms):
1. Early Warning Signs (Infants)
- Delayed milestones (Sitting, Crawling, Walking).
- Poor head control (After 3 months).
- Abnormal muscle tone (Too stiff or floppy).
- Persistent primitive reflexes (Moro, Tonic Neck Reflex beyond 6 months).
2. Motor & Postural Abnormalities (Main Features)
- Spasticity (Stiff, tight muscles) – Seen in Spastic CP.
- Involuntary movements (Tremors, Writhing motions) – Seen in Dyskinetic CP.
- Uncoordinated walking (Gait abnormalities, Toe-walking, Scissor gait).
- Speech difficulties (Dysarthria, Drooling).
3. Additional Complications
- Intellectual disability (50% of cases).
- Seizures (Common in Spastic CP).
- Hearing & Vision Impairments.
- Feeding difficulties (Dysphagia, Malnutrition).
Diagnosis:
1. Developmental Screening Tests
- Ages and Stages Questionnaire (ASQ).
- Denver Developmental Screening Test (DDST-II).
2. Neurological Examination
- Muscle tone, Reflexes, Postural assessment.
- Persistence of primitive reflexes (e.g., Babinski, Moro reflex beyond infancy).
3. Imaging Studies
- MRI Brain – Identifies brain abnormalities (White matter damage, Periventricular leukomalacia).
- CT Scan – Detects brain malformations or calcifications.
4. Electroencephalogram (EEG)
- If seizures are present.
5. Genetic & Metabolic Tests
- For suspected hereditary or metabolic disorders.
Medical Management (Non-Surgical Treatment):
1. Medications to Control Muscle Spasticity
- Baclofen (Oral or Intrathecal pump) – Reduces muscle tightness.
- Diazepam (Benzodiazepine) – For muscle relaxation.
- Botulinum Toxin (Botox) Injections – Temporary muscle relaxation.
2. Seizure Management
- Antiepileptic drugs (Valproate, Levetiracetam, Carbamazepine).
3. Pain Management
- NSAIDs, Acetaminophen (For joint pain & contractures).
4. Speech & Swallowing Therapy
- Speech therapy for communication improvement.
- Gastrostomy tube (G-tube) feeding for severe dysphagia.
5. Occupational & Physical Therapy
- Stretching, Strengthening, Balance training.
- Use of orthotic devices (Braces, Walkers, Wheelchairs).
Surgical Management:
1. Orthopedic Surgeries (For Mobility & Posture)
- Tendon release surgery (For tight hamstrings, Achilles tendons).
- Hip reconstruction (For hip dislocation).
- Scoliosis correction (For spinal deformities).
2. Selective Dorsal Rhizotomy (SDR)
- Surgical cutting of specific nerve roots to reduce spasticity.
3. Deep Brain Stimulation (DBS)
- Used in severe Dyskinetic CP (Basal ganglia dysfunction).
Nursing Management:
1. Early Intervention & Developmental Support
- Monitor growth, weight, and nutrition status.
- Encourage early physiotherapy & mobility exercises.
2. Seizure Precautions & Safety
- Ensure safe environment (Padding, Helmet if frequent falls).
- Administer anticonvulsants as prescribed.
3. Nutritional & Feeding Support
- Encourage high-calorie diet (Prevent malnutrition).
- Assist with feeding (Use adaptive feeding tools).
4. Preventing Contractures & Deformities
- Encourage passive stretching exercises.
- Use braces & splints as needed.
5. Emotional & Social Support
- Encourage school integration, social interactions.
- Support parents in managing emotional & financial burdens.
Complications of Cerebral Palsy:
- Muscle contractures → Joint deformities.
- Aspiration pneumonia (Due to difficulty swallowing).
- Seizures & Cognitive Impairment.
- Chronic pain & Fatigue.
- Psychosocial challenges (Depression, Low self-esteem).
Prognosis:
- Mild CP → Good prognosis, independent mobility.
- Severe CP → Requires lifelong care & assistance.
- Early interventions improve quality of life & functional independence.
Key Points:
✔ Cerebral Palsy is a non-progressive motor disorder affecting movement & posture.
✔ Common causes include birth asphyxia, infections, prematurity.
✔ Diagnosis involves MRI, developmental assessments, neurological exams.
✔ Treatment focuses on physical therapy, muscle relaxants, orthopedic surgeries.
✔ Nursing care includes mobility support, nutritional assistance, and emotional support
Head Injury in Children
Definition:
Head injury in children refers to trauma to the scalp, skull, or brain resulting in neurological dysfunction, ranging from mild concussion to severe brain damage. It can lead to intracranial bleeding, skull fractures, or long-term cognitive and motor impairments.
Etiology (Causes & Risk Factors):
1. Common Causes of Head Injury in Children
- Falls (Most Common) – From beds, stairs, playgrounds.
- Motor vehicle accidents – Bicycle crashes, Car crashes (Without seatbelt use).
- Child abuse (Shaken baby syndrome, Blunt trauma).
- Sports injuries – Football, Boxing, Gymnastics.
- Penetrating injuries – Gunshot wounds, Sharp objects.
2. Risk Factors
- Young age (<5 years) – Due to poor coordination & weak neck muscles.
- Lack of safety measures (No helmet, No seatbelt).
- High-impact activities without protective gear.
- Neurological disorders (Seizures, ADHD – Higher risk of falls).
Types of Head Injury:
| Type | Description | Clinical Features |
|---|---|---|
| Concussion (Mild TBI) | Temporary brain dysfunction due to trauma | Headache, Confusion, Dizziness, Vomiting |
| Contusion (Brain Bruise) | Localized bleeding in the brain | Neurological deficits, Drowsiness, Seizures |
| Skull Fracture | Break in the skull bone | Pain, Swelling, CSF leakage from nose/ears |
| Epidural Hematoma | Bleeding between skull & dura mater | Loss of consciousness → Brief recovery → Rapid deterioration |
| Subdural Hematoma | Bleeding between dura & arachnoid mater | Gradual worsening of consciousness, Seizures |
| Diffuse Axonal Injury (DAI) | Widespread brain damage due to shearing forces | Immediate coma, Brain swelling, Poor prognosis |
Pathophysiology:
- Primary Brain Injury (Immediate Impact)
- Direct trauma causes damage to brain cells, blood vessels, or neurons.
- Leads to cerebral contusion, concussion, or skull fractures.
- Secondary Brain Injury (Delayed Effects)
- Cerebral edema → Brain swelling increases intracranial pressure (ICP).
- Hypoxia & Ischemia → Decreased oxygen supply worsens brain damage.
- Intracranial bleeding (Hematomas) → Further compresses brain structures.
- Intracranial Pressure (ICP) Elevation
- Increased pressure inside the skull leads to brain herniation if untreated.
Clinical Manifestations (Signs & Symptoms):
1. Mild Head Injury (Concussion)
- Headache, Dizziness, Confusion.
- Loss of consciousness (<30 minutes).
- Vomiting, Irritability.
- Amnesia (Forgetting the event).
2. Moderate to Severe Head Injury
- Prolonged loss of consciousness (>30 minutes).
- Repeated vomiting.
- Seizures, Weakness in limbs.
- Slurred speech, Inability to wake up.
3. Signs of Increased Intracranial Pressure (ICP)
- Cushing’s Triad (Hypertension, Bradycardia, Irregular breathing).
- Unequal pupil size (Anisocoria).
- Bulging fontanelle (In infants).
- Papilledema (Swelling of the optic nerve, Blurred vision).
Diagnosis:
1. Glasgow Coma Scale (GCS) – Severity Assessment
| Score | Severity | Response |
|---|---|---|
| 13-15 | Mild Head Injury (Concussion) | Awake, Oriented, Minor symptoms |
| 9-12 | Moderate Head Injury | Confused, Lethargic, Some motor weakness |
| <8 | Severe Head Injury | Unconscious, Requires airway support |
2. Imaging Studies
- CT Scan of Brain – Gold Standard for Head Injury
- Detects intracranial bleeding, skull fractures, brain swelling.
- MRI Brain (For Diffuse Axonal Injury, Chronic Injury).
3. Laboratory Tests
- Blood Glucose – Hypoglycemia can worsen brain injury.
- Coagulation Profile – To detect bleeding disorders.
- Serum Electrolytes – Hyponatremia can cause cerebral edema.
Medical Management (Non-Surgical Treatment):
1. Emergency Management (For Severe Cases)
- Airway Protection (Intubation if GCS <8).
- Oxygen therapy (Maintain brain oxygen supply).
- IV Fluids – To prevent hypotension, but avoid fluid overload.
- Elevate head (30 degrees) to reduce ICP.
2. Medications
- Mannitol (Osmotic Diuretic) – Reduces brain swelling.
- Hypertonic Saline – Used in severe brain edema.
- Anticonvulsants (Phenytoin, Levetiracetam) – For seizure prevention.
- Pain Control (Acetaminophen, Avoid NSAIDs due to bleeding risk).
- Antibiotics (For open skull fractures or penetrating injuries).
3. Monitoring & Rehabilitation
- Frequent neurological checks (GCS, Pupillary response).
- Speech & Occupational Therapy (For cognitive rehabilitation).
Surgical Management (For Severe Brain Injury):
1. Craniotomy (For Bleeding & Brain Swelling)
- Removes hematomas (Epidural/Subdural Hematoma).
- Relieves pressure inside the brain.
2. Decompressive Craniectomy
- Removes part of the skull to allow brain swelling to expand outward.
- Used in severe traumatic brain injury (TBI) with cerebral edema.
3. Ventriculostomy (External Ventricular Drain – EVD)
- Used to drain excess cerebrospinal fluid (CSF) in hydrocephalus.
Nursing Management:
1. Immediate Post-Injury Care
- Assess airway, breathing, circulation (ABC).
- Monitor vital signs & neurological status every 15-30 minutes.
- Keep head elevated to reduce ICP.
- Avoid excessive stimulation (Keep environment quiet).
2. Post-Surgical Nursing Care
- Monitor for signs of infection (Fever, CSF leakage from wound).
- Assess for worsening neurological status (Seizures, Weakness).
- Provide psychological support to parents.
3. Long-Term Nursing Care & Rehabilitation
- Physical therapy – Improve mobility & prevent contractures.
- Cognitive rehabilitation – Improve memory, speech.
- Educate family on home safety (Helmet use, Fall prevention).
Complications of Head Injury:
- Post-Concussion Syndrome (Headaches, Dizziness, Memory issues).
- Seizures (Post-Traumatic Epilepsy).
- Chronic Traumatic Encephalopathy (Repeated head injuries).
- Hydrocephalus (CSF accumulation, requiring VP Shunt).
- Brain Herniation (If untreated ICP increases).
Prognosis:
- Mild Head Injury (Concussion) → Good prognosis, full recovery.
- Moderate Head Injury → Recovery possible, may have some cognitive impairments.
- Severe Head Injury → Risk of long-term disability, requires rehabilitation.
Key Points:
✔ Head injury in children ranges from mild (concussion) to severe (TBI).
✔ Common causes include falls, vehicle accidents, child abuse.
✔ Diagnosis involves CT Scan, GCS assessment, neurological exams.
✔ Treatment includes ICP management, seizure control, and rehabilitation.
✔ Nursing care focuses on airway protection, neuro monitoring, and family education.
Juvenile Diabetes Mellitus (Type 1 Diabetes in Children)
Definition:
Juvenile Diabetes Mellitus, also known as Type 1 Diabetes Mellitus (T1DM), is a chronic autoimmune disorder characterized by destruction of insulin-producing beta cells in the pancreas, leading to absolute insulin deficiency. It requires lifelong insulin therapy and primarily occurs in children and adolescents.
Etiology (Causes & Risk Factors):
1. Autoimmune Destruction (Most Common Cause)
- Genetic predisposition (HLA-DR3, HLA-DR4 genes).
- Autoantibodies (Islet cell antibodies, Anti-GAD antibodies) attack pancreatic beta cells.
2. Environmental Triggers
- Viral infections (Coxsackievirus, Enteroviruses, Mumps, Rubella).
- Early exposure to cow’s milk proteins (In susceptible infants).
- Vitamin D deficiency (May increase risk).
3. Genetic Factors
- Family history of Type 1 Diabetes (Siblings or Parents affected).
4. Other Risk Factors
- Stress, Obesity (Increases insulin resistance).
- Certain autoimmune disorders (Hashimoto’s Thyroiditis, Celiac Disease).
Types of Diabetes in Children:
| Type | Cause | Features |
|---|---|---|
| Type 1 Diabetes Mellitus (T1DM) | Autoimmune destruction of beta cells | Insulin-dependent, Early onset (Usually <20 years) |
| Type 2 Diabetes Mellitus (T2DM) | Insulin resistance, Beta cell dysfunction | Obesity-related, Insulin resistance, Increasing in teens |
| Maturity-Onset Diabetes of the Young (MODY) | Genetic mutations in beta cells | Mild, Runs in families, May not need insulin |
Pathophysiology of Type 1 Diabetes Mellitus (T1DM):
- Autoimmune Attack on Pancreatic Beta Cells
- Cytotoxic T-cells & Autoantibodies destroy insulin-producing beta cells in the pancreas.
- Complete Insulin Deficiency Develops
- Glucose cannot enter cells → Blood glucose levels rise (Hyperglycemia).
- Compensatory Mechanisms Triggered:
- Glycogen breakdown (Glycogenolysis) → More glucose enters the bloodstream.
- Fat breakdown (Lipolysis) → Produces ketones → Leads to diabetic ketoacidosis (DKA).
- Glucose Spills into Urine (Glycosuria):
- Osmotic diuresis → Leads to dehydration, Polyuria, Polydipsia.
Clinical Manifestations (Signs & Symptoms):
1. Classic Symptoms (“3 P’s”)
- Polyuria (Excessive urination) → Due to osmotic diuresis.
- Polydipsia (Excessive thirst) → Due to dehydration.
- Polyphagia (Excessive hunger) → Due to lack of glucose in cells.
2. Other Common Symptoms
- Unexplained weight loss (Fat breakdown for energy).
- Fatigue, Weakness.
- Blurred vision (Due to fluid shifts in the eye lens).
- Frequent infections (UTIs, Skin infections).
- Fruity breath odor (Due to ketone production).
3. Signs of Diabetic Ketoacidosis (DKA) – Emergency
- Severe dehydration.
- Rapid, deep breathing (Kussmaul’s respirations).
- Nausea, Vomiting, Abdominal pain.
- Altered mental status (Confusion, Drowsiness, Coma).
Diagnosis:
1. Blood Tests:
| Test | Normal Range | Diabetes Diagnosis |
|---|---|---|
| Fasting Blood Glucose | <100 mg/dL | ≥126 mg/dL |
| Random Blood Glucose | <140 mg/dL | ≥200 mg/dL + Symptoms |
| HbA1c (Glycated Hemoglobin) | <5.7% | ≥6.5% |
| Oral Glucose Tolerance Test (OGTT – 2hr Value) | <140 mg/dL | ≥200 mg/dL |
2. Autoimmune Markers (For Type 1 Diabetes)
- Islet Cell Antibodies (ICA).
- Glutamic Acid Decarboxylase (GAD) Antibodies.
3. Urine Tests:
- Glycosuria (Glucose in urine).
- Ketonuria (Ketones in urine – Indicates DKA).
Medical Management (Non-Surgical Treatment):
1. Insulin Therapy (Lifelong Treatment)
| Type of Insulin | Onset | Peak | Duration | Examples |
|---|---|---|---|---|
| Rapid-Acting | 15 min | 1-2 hrs | 3-5 hrs | Lispro, Aspart |
| Short-Acting | 30 min | 2-4 hrs | 5-8 hrs | Regular Insulin |
| Intermediate-Acting | 1-2 hrs | 4-12 hrs | 12-24 hrs | NPH |
| Long-Acting | 2-4 hrs | None | 24 hrs | Glargine (Lantus), Detemir |
- Basal-Bolus Regimen (Best for children).
- Insulin Pump Therapy – Continuous delivery for better glucose control.
2. Blood Glucose Monitoring
- Frequent glucose checks (Before meals & bedtime).
- Target Blood Glucose: 80-150 mg/dL.
3. Nutritional Management
- Carbohydrate counting (Insulin dosing based on carb intake).
- Balanced diet with complex carbs, protein, and healthy fats.
- Avoid sugary drinks, processed foods.
4. Exercise Management
- Regular physical activity improves insulin sensitivity.
- Monitor glucose before & after exercise to prevent hypoglycemia.
5. Hypoglycemia Treatment (If Glucose <70 mg/dL)
- 15g of fast-acting sugar (Juice, Glucose tablets).
- Glucagon injection if unconscious.
Surgical Management:
- Pancreatic Islet Cell Transplantation (Experimental Therapy).
- Pancreas Transplant (Rarely done in children).
Nursing Management:
1. Acute Care (During Diagnosis or DKA)
- Monitor vital signs & neurological status (Risk of coma in DKA).
- Administer IV fluids (For dehydration).
- Administer insulin therapy as prescribed.
- Monitor blood glucose & ketones hourly.
2. Long-Term Nursing Care
- Educate family about insulin administration & glucose monitoring.
- Reinforce healthy eating & exercise habits.
- Encourage school accommodations (Diabetes action plan).
- Monitor for complications (Foot care, Eye exams).
Complications of Juvenile Diabetes:
1. Acute Complications
- Diabetic Ketoacidosis (DKA) – Medical emergency.
- Hypoglycemia (Shakiness, Sweating, Seizures, Coma).
2. Chronic Complications
- Retinopathy (Eye damage).
- Nephropathy (Kidney disease).
- Neuropathy (Nerve damage).
- Delayed wound healing (Risk of infections).
Prognosis:
- Well-managed Type 1 Diabetes allows a normal, healthy life.
- Poor control leads to severe complications in adulthood.
- Lifelong insulin therapy & self-care education are essential.
Key Points:
✔ Juvenile Diabetes is an autoimmune disorder causing insulin deficiency.
✔ Symptoms include excessive thirst, urination, weight loss, fatigue.
✔ Diagnosis involves blood glucose tests, HbA1c, and autoantibody screening.
✔ Treatment includes insulin therapy, blood sugar monitoring, and lifestyle changes.
✔ Nursing care focuses on education, glucose control, and preventing complications.