ENGLISH PERDIATRIC UNIT 5 CONG. GI. SYSTEM.
Children with Congenital Disorder of the Gastro intestinal (GI) system.
Explain/ Define Cleft lip and Cleft palate
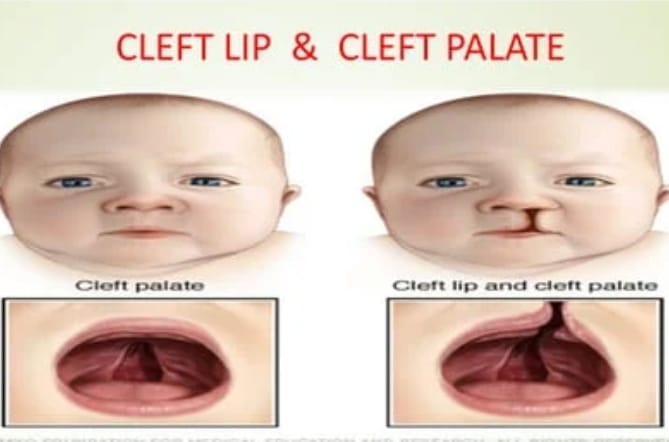
Cleft lip and cleft palate are congenital malformations of the face caused by failure of fusion of the first brachial arch during intrauterine development.
Cleft lip and cleft palate are usually seen due to failure of development of bony and soft tissue structures on one or both sides of the midline of the palate and upper jaw.
Complete formation of the lip takes place between 5 and 12 weeks during intrauterine life.
While the complete formation of the palette takes place during 12 to 14 weeks.
Cleft lip and cleft palate can also occur with other congenital malformations such as central nervous system (CNS) anomalies, cardiovascular system anomalies and any congenital anomalies of the skeletal system.
Cleft lip and cleft palate can occur single or in combination and are seen unilaterally and bilaterally.
Cleft lip (cheiloschisis)
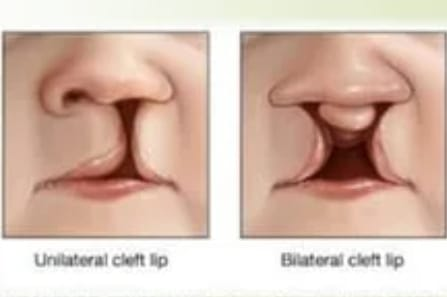
Cleft lip is a congenital anomaly of the face that results in a gap between the lips due to failure of fusion of the medial tissues with the lateral tissues that normally form the upper lip, with separation and opening of the upper lip.
A cleft lip usually extends from a small dimple to a large gap or sometimes even to the nose. The condition of cleft lips usually occurs during early fetal development or when the tissues of the lips are forming due to lack of proper fusion, leaving a gap between the lips and the condition of cleft lips arises.
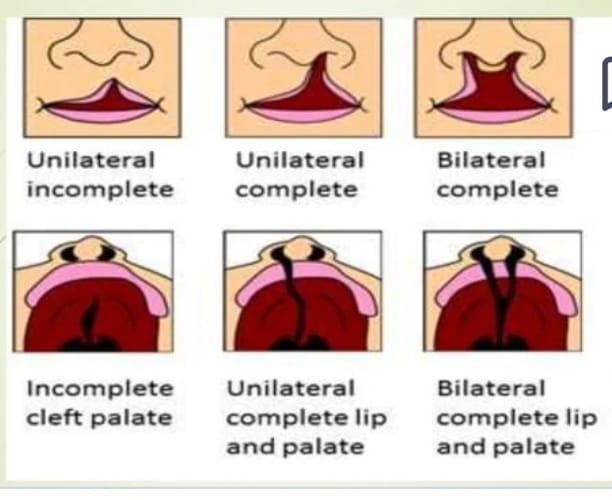
type
Partial and Incomplete,
complete,
Unilateral,
Bilateral.
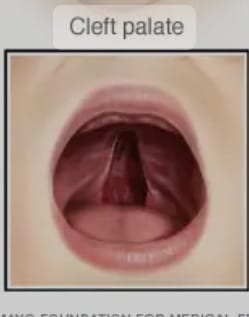
Cleft palate (palatoschisis)
Cleft palate is usually a congenital anomaly of the palate. A cleft palate is a condition in which there is a gap and opening in the roof of the mouth.
A condition of left palate is usually left during the development of the palette during intrauterine life due to the failure of the secondary palette to fuse properly with each other and with the primary palette and the tissues that form the palette. Arises.
Which can affect the front, back or both patties of the pallet.
type
complete,
Incomplete.
Explain the Etiology/cause of the child with the cleft lip and cleft palate
Due to genetic factors,
Due to any type of infection in the mother between 5 to 12 weeks during pregnancy,
Due to certain environmental factors,
Due to mother’s exposure to any teratogenic substance such as smoking, alcohol, and consumption of certain types of medicine (anti-convulsive, chemotherapy drug, methotrexate drug) during pregnancy.
Due to non-nutritious diet intake by the mother during pregnancy,
Due to exposure to any chemicals and radiation while the mother is pregnant,
Any other medical condition in the mother like anemia, hypoproteinemia etc.
Due to the mother having nutritional problems during pregnancy,
Due to impaired vascular supply to the fetus.
Explain the clinical manifestation/sign and symptoms of the child with the cleft lip and cleft palate
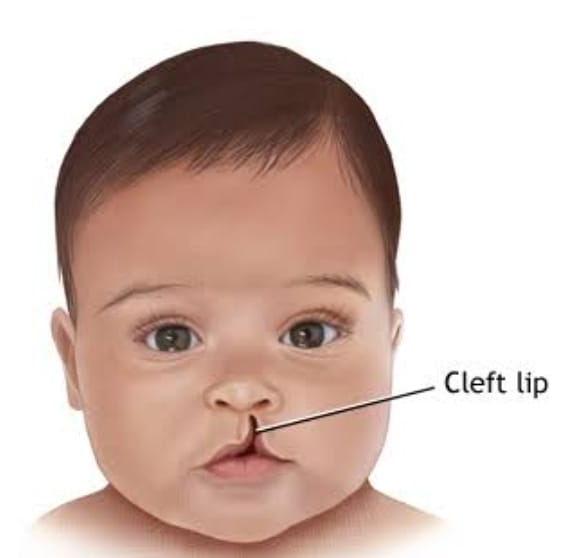
Having a visible cleft clip,
Seeing the left palette,
Feeding difficulties,
Having speech difficulties,
Impairment in pronouncing certain types of sounds,
Dental problems,
Ear infection and hearing problem,
Nasal congestion,
Breathing difficulties,
Explain the Diagnostic evaluation of the child with the cleft lip and cleft palate
- History taking and physical examination,
- Complete physical examination at birth,
- imaging studies,
- ultrasound,
- x ray,
- MRI,
- Hearing Assessment,
- Feeding Assessment,
- speech evaluation,
- dental evaluation,
- Genetic assessment.
Explain the Complication of the child with the cleft lip and cleft palate
- feeding difficulties,
- getting an ear infection,
- Hearing problem,
- Speech problem,
- Malocclusion of teeth and mal placement,
- Abnormalities in facial and dental growth,
- Recurrent infections, especially otitis media,
- Nasal issues especially breathing difficulties,
- Feeding difficulties and speech problems,
- Alteration of body image.
Explain the surgical management of the child with the cleft lip and cleft palate
In the surgical management of cleft lip and cleft palate, first the cleft lip is surgically corrected and then the cleft palate is surgically repaired.
A cleft lip is surgically repaired two to three months after birth.
Cleft lip is surgically repaired in a Z shape.
After surgically repairing the left lips, the sutures are protected with this metal device.
After the left lip is surgically repaired, the cleft palate is surgically repaired. This procedure is usually performed on children around the age of 1 to 2 years.
A child who has a condition of cleft palate has a speech problem, so to correct the same speech problem, proper speech therapy should be provided to the child.
Explain the nursing management of the child with the cleft lip and cleft palate
Airway clearance
Providing proper upright position to the child, providing proper hand support to the child’s head, thereby reducing the possibility of aspiration and maintaining proper airway clearance of the child.
Feeding support
Conditions like cleft palate and left palate cause shaking problems in the child due to which proper feeding should be provided. Use of special cleft nipple to maintain feeding condition of child. Use of special type of bottle and feeding device.
Hearing monitoring
If the child has the condition of cleft palate, there is a possibility of middle ear infection. So assess the child’s hearing ability.
Speech therapy
A child with a condition of cleft palate has speech difficulties so proper speech therapy should be provided to the child.
Dental care
If the child has the condition of cleft lip and left palate, there is a possibility of teeth malplacement, so the child should be provided with proper dental care.
Psychological support
If the parents of the child with left clip and cleft palate condition are emotionally disturbed, then proper psychological support, counselling, emotional support and social support should be provided to the parents.
Monitoring and follow up
Advising the parents to take regular follow up for proper care of the child.
Preoperative nursing management
Follow the “Rule of Ten” before surgery on a child with a left clip.
(10 wick age, 10 lbs weight, 10 gm% of hb).
If the child has a cleft palate then 12 months, weight is 9 kg(20 lbs) and 10 gm Hb %.
Child should be fully immunized before surgery.
If the child has a habit of thumb shaking, give advice to prevent his thumb shaking.
Advising mothers on sterile breastfeeding techniques.
Advising the mother to provide proper best feeding to her child.
Providing a proper position to the child for the comfort level of the child.
Giving advice to his child to provide proper love and affection to the child.
Postoperative nursing management
To continuously monitor the child’s vital signs.
To provide general post operative care to the child.
Provide proper side lining position to drain child’s secretions and prevent aspiration.
Providing proper protection to the child’s surgical site.
To use a properly protective device on the sutures side.
Provide the child with proper supine position and position on the unaffected side.
Provide proper elbow restraint to the child.
Giving advice to the parents of the child to provide proper love and affection to the child and giving advice to the parents to provide support to the child.
Advise the child to provide feeding through medicine dropper properly.
Advise the parents of the child to maintain proper upright position of the child after providing feeding.
Advise the child to maintain aseptic technique to prevent infection.
Advise the parents to maintain proper hygiene while handling the child.
Advising on providing proper assistance when providing child care.
If possible, advise the child’s parents to use a special cleft palate nipple.
Giving advice to parents to provide proper love and affection to their child.
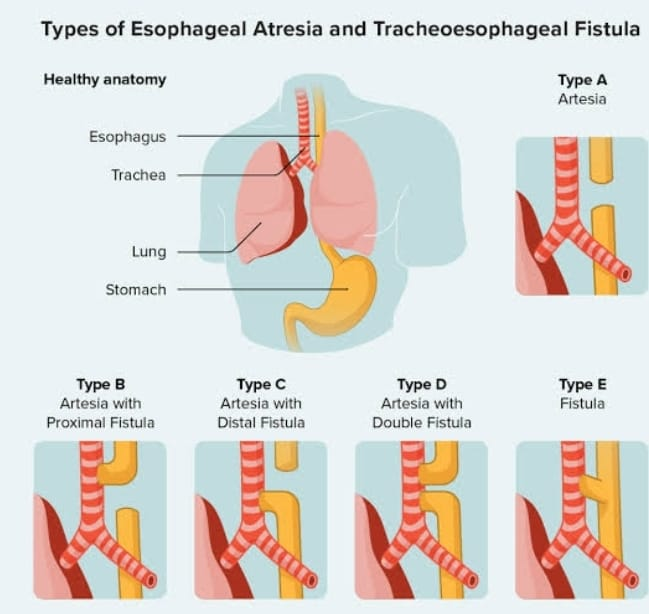
- Explain/Define Esophageal Atresia Tracheoesophageal fistula (TEF) (
Esophageal atresia
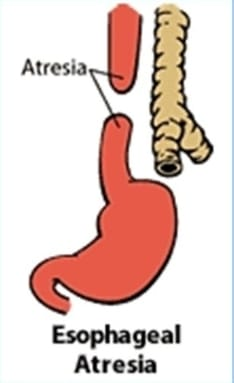
Esophageal atresia is a congenital anomaly of the esophagus. In which the esophagus does not develop properly.
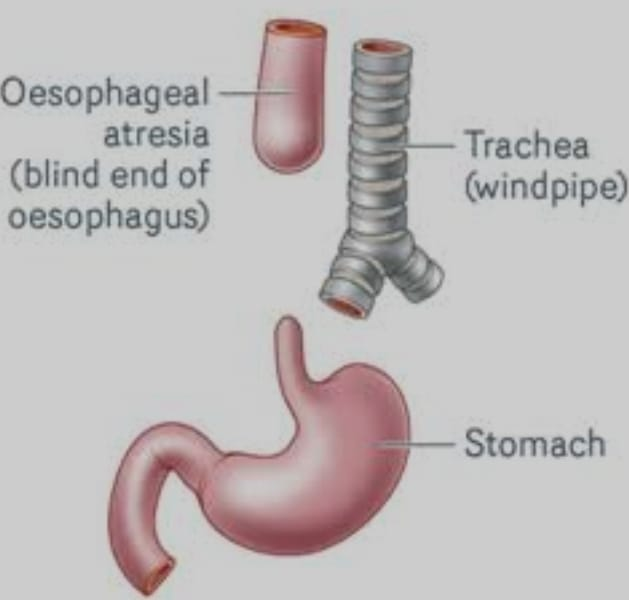
Esophageal atresia is a condition of the esophagus in which there is a failure of the formation of a continuous passage from the pharynx of the esophagus to the stomach during embryonic development. That is, the esophagus is closed at one or more places or a part of the esophagus is absent. It causes discontinuity of esophagus and formation of gap between it and due to which food and liquid cannot reach from mouth to stomach.
Tracheoesophageal Fistula (TEF)
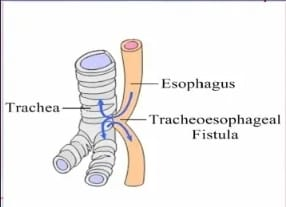
A tracheoesophageal fistula is an abnormal communication between the esophagus (the tube that is responsible for passing food from the mouth to the stomach) and the trachea (the tube that carries air to the lungs and works to take air from the lungs).
Tracheoesophageal fistula is a congenital anomaly in which there is an abnormal communication/connection between the esophagus and the trachea.
This is a type of congenital disorder that is usually seen in premature children, low birth weight and in children whose mother has the condition of polyhydroamnios.
Congenital heart disease and gastro-intestinal (GI) anomalies are also seen in children with this tracheoesophageal fistula condition.
Explain the Etiology/cause of the child with the Tracheoesophageal fistula.
- The exact cause of this condition is unknown.
- Due to genetic factors,
- Due to environmental factors,
- If the mother smokes, consumes alcohol and takes certain types of drugs during pregnancy,
- Due to certain maternal health conditions like diabetes, obesity etc.
- Due to maternal polyhydroamnios,
- teratogenic stimuli,
- Due to the intra-uterine environment,
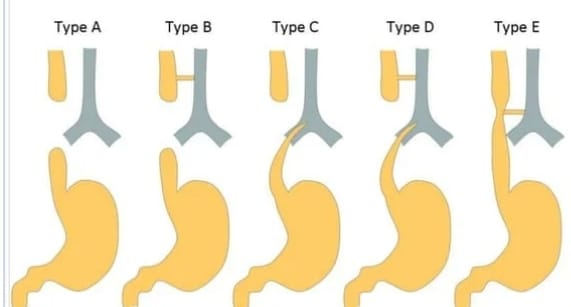
Explain the types of the Tracheoesophageal fistula
There are five types of tracheoesophageal fistula.
Type A := Without fistula
In this type, there is no communication between the esophagus and the trachea, it has a blind pouch and the upper segment (proximal) of the esophagus is a blind pouch and its lower segment (distal) is also a blind pouch.
Type B – Tracheoesophageal fistula (upper segment)
In this type, the upper segment (proximal) of the esophagus forms a fistula (abnormal communication) with the trachea, while the lower segment (distal) of the esophagus is a blind pouch.
Type c – tracheoesophageal fistula (lower segment)
In this type, the upper segment of the esophagus (proximal) is a blind pouch while the lower part (distal) of the esophagus forms a fistula with the trachea.
Type D – Tracheoesophageal fistula (upper segment and lower segment communication)
In this type, the upper part and lower part of the esophagus have an abnormal communication with the trachea.
Type E – H Type
In this type there is no blind pouch of esophagus but it has abnormal communication with trachea with formation of H shape tracheoesophageal fistula.
Explain the clinical manifestation/sign and symptoms of the child with the Tracheoesophageal fistula
- Excessive drooling,
- Excessive salivation,
- When an infant or neonate a
- While trying to feed, cuffing, choking and sneezing, sometimes fluid enters the trachea.
- passing secretions in large amounts from the nose,
- When the child is feeding, he swallows but passes out by vomiting and coughing.
- Bluish discoloration in the skin,
- Breathing difficulty,
- Abdominal distension,
- regurgitation,
- Failure to thrive,
- Having excessive salivation in the mouth,
- Having recurrent respiratory infections,
- Respiratory distress,
- Poor feeding.
Explain the Diagnostic evaluation of the child with the Tracheoesophageal fistula
- History taking and physical examination,
- imaging studies,
- ultrasonography,
- chest x-ray,
- contrast esophagography,
- bronchoscopy,
- Assessing the blind pouch by inserting a radiopaque catheter.
Explain the medical management of the child with the Tracheoesophageal fistula
As soon as the diagnosis of tracheoesophageal fistula is made, provide the child with proper upright position.
Properly stabilize the child and provide respiratory support.
To improve the child’s breathing condition by providing him with proper oxygen.
A child with a condition of tracheoesophageal fistula cannot take oral feeding, so insertion of a nasogastric tube can be done to provide them with fluid.
Provide intravenous fluid to the child properly.
Avoiding a child with a nasogastric tube inserted can prevent aspiration.
Keeping the child on NPO until the child’s condition is treated surgically can prevent aspiration.
Provide proper intravenous fluids to maintain child’s fluid and electrolyte levels.
If the child is likely to get any infection, provide proper antibiotic medication.
To provide complete education to the child’s parents about the child’s condition and to provide them with psychological support and counselling.
Explain the Surgical management of the child with the Tracheoesophageal fistula
Surgical correction of a child with tracheoesophageal fistula by anastomosis usually depends on the distance between the upper blind pouch and the lower blind pouch, the type of defect, the condition of the neonate, its weight and the severity of the defect.
First perform a gastrostomy to prevent aspiration.
If the distance between upper blind pouch and lower blind pouch is less than 2.5 cm then tracheostomy is performed and tracheoesophageal fistula is divided and ligation is done and then anastomosis is performed between upper segment and lower segment of esophagus.
If the distance between the two pouches is greater and the condition of the child is poor, the procedure is performed in two stages.
Stage 1 := In this stage esophageal fistula is ligated and gastrostomy is performed.
Stage 2 := In this stage at the same time of 18 months the distance is completed using the colon segment after which the esophagostomy and gastrostomy are closed.
Explain the Nursing management of the child with the Tracheoesophageal fistula
Preoperative nursing management
Early detection of child’s condition.
After diagnosing the condition, providing the child with a properly upright position to prevent aspiration.
Perform exercises frequently.
Keep the child on oral (NPO) to reduce respiratory infections.
Keeping the child’s airways patent and providing oxygen properly.
To continuously monitor the child’s whiter signs.
Provide appropriate intravenous fluids to maintain the child’s electrolyte levels and prevent dehydration.
Properly maintain intake output chart of child.
Continuously monitoring the health status of the child.
To assess whether the child has the condition of abdominal distension or not.
Complete education of the child’s parents about the child’s condition and provide proper emotional support to reduce his anxiety.
Postoperative nursing management
Keeping the child’s airway properly patent.
Performing a proper examination.
Monitoring thoracic drainage.
Provide proper oxygen support to the child.
To properly monitor the child’s vital signs.
Provide proper intravenous fluids to the child.
Provide proper elbow restraint to the child.
If the child is in pain, provide proper analgesic medication.
To provide proper comfort measures to the child.
Take necessary precautions to continuously monitor chest tube drainage.
To prevent the child from infection, maintain aseptic technique, provide proper dressing and maintain the child’s hygienic and cleanliness properly.
Administering proper antibiotic medication to prevent the child from infection.
Continuously monitoring the child’s condition.
To provide education to the child’s parents and their family members to take proper care of the child.
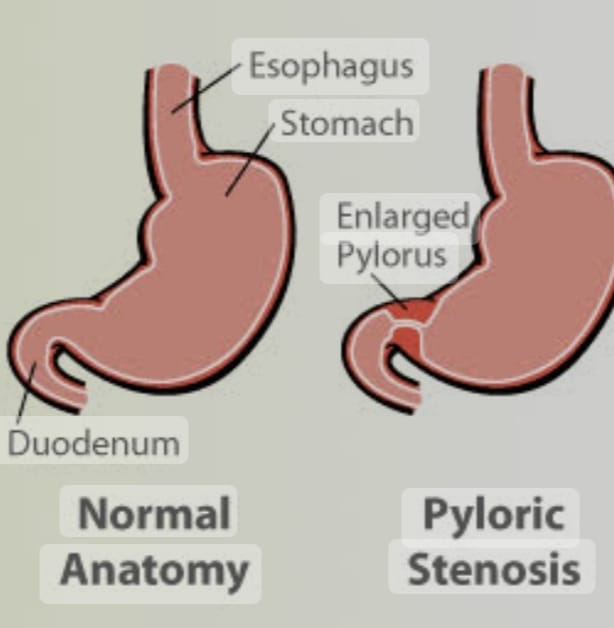
- Explain/Define Congenital Hypertrophy pyloric Stenosis
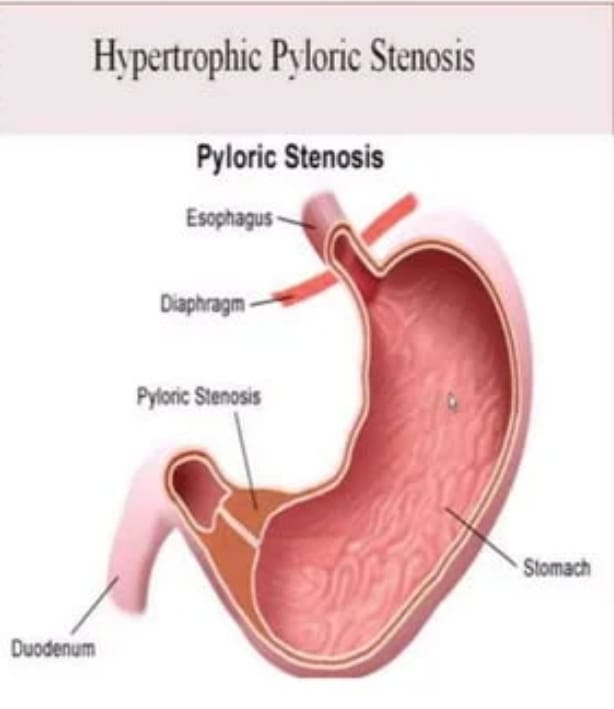
Hypertrophy: = thickening of muscle fibers.
Pyloric := Pyloric tract of stomach.
Stenosis: = Means narrowing of body part.
Narrowing of pyloric band due to thickening of muscle fibers of pyloric band of mince stomach.
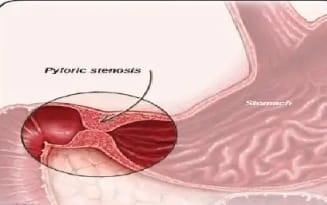
Congenital hypertrophic pyloric stenosis (CHPS) is a condition seen in infants where the muscles around the outlet of the stomach thicken.
In congenital hypertrophic pyloric stenosis, there is progressive thickening, overgrowth and enlargement of the circular muscle fibers of the pylorus portion of the stomach, resulting in partial and total obstruction of the outlet of the stomach and narrowing of the pyloric portion, thus blocking the passage of food into the small intestine. happens
This results in symptoms such as projectile vomiting, weight loss, and dehydration, which usually occur during the first few weeks of life. It usually requires surgical intervention to correct it.
Explain the Etiology/cause of the pyloric Stenosis.
- EXIT CAUSE IS UNKNOWN,
- Due to genetic factors,
- Due to environmental factors,
- Due to mother taking stress in last trimester,
- Due to elevated prostaglandin levels,
- Nitric acid deficiency,
- Due to mother’s smoking during pregnancy,
- Due to family history,
- Ethnic origin (mainly in white),
- It is more common in boys than girls (7:1).
- Due to pre-mature birth,
- Due to providing some type of antibiotic medication to the infant in the first week of birth, such as erythromycin,
- Because of bottle feeding.
Explain the Clinical manifestation/ sign and symptoms of the pyloric Stenosis.
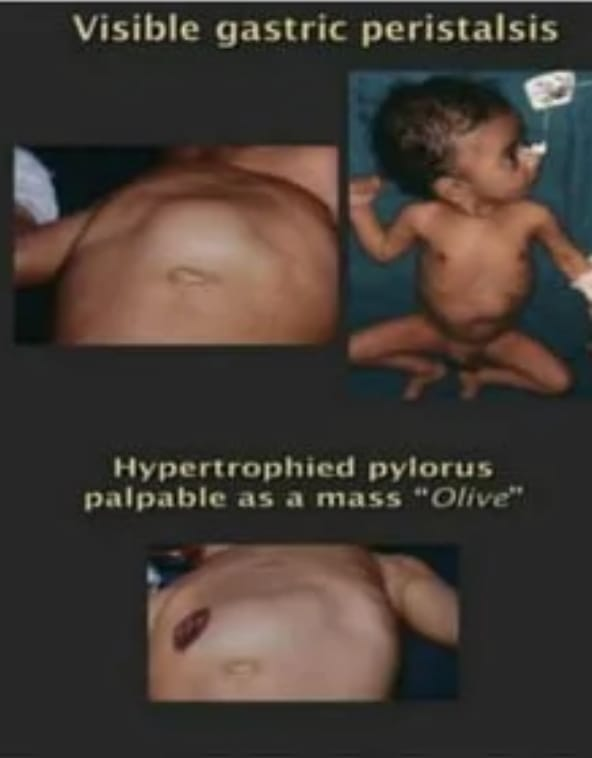
- A palpable olive sap mass like structure is felt in the upper right quadrant,
- Projectile vomiting after feeding,
- weight loss,
- Dehydration,
- dry mouth,
- Constriction of fontanelles,
- Decreased urination,
- Frequency and intensity of vomiting gradually increase,
- Peristalsis moment visible from left side to right side after baby feeding
- A child is continuously hungry,
- Irritability,
- Condition of Failure to Thrive,
- The child appears lethargic,
- Respiration becomes sluggish,
- Malnutrition,
- Constipation.
Explain the Diagnostic evaluation of the pyloric Stenosis.
- History taking and physical examination,
- Plain X Ray Abdomen,
- ultrasonography,
- Note the nature and type of vomiting,
- A palpable mass felt at the pyloric sphincter,
- barium x ray,
- Blood Examination for HB%,
- Blood PH Test,
- Serum electrolyte and urine examination,
- Arterial blood gas analysis.
Explain the management of the pyloric stenosis.
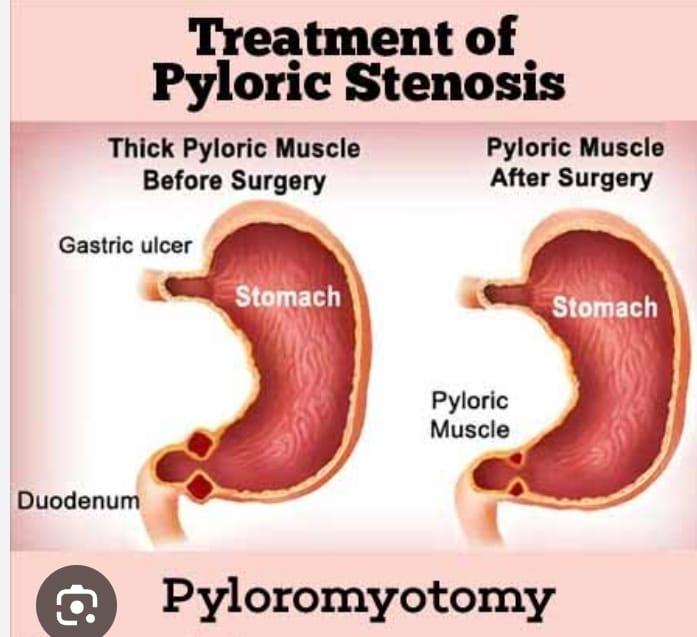
Fredet-Ramsteds procedure
The Fredet-Ramstedt procedure, also known as pyloromyotomy, is a surgical procedure used to treat congenital hypertrophic pyloric stenosis (CHPS). It was developed independently in the late 19th century by surgeons Friedrich Ramstedt and Pierre Fredet. In this procedure:
1.Incision:
An incision is made along the longitudinal axis of the hypertrophied pyloric muscles, especially on the anterior surface of the stomach.
2.Muscle Division:
without damaging the mucosa (inner lining) of the stomach or the underlying blood vessels
The thick muscle of the pylorus is carefully divided along its length.
- Widening of pyloric channel:
By dividing the hypertrophoid muscles, the part between the stomach and the small intestine (pyloric tract) is widened, allowing food to enter the small intestine normally. - Closure:
After division of the muscles, the incision is closed with sutures.
The Fredet-Ramstedt procedure is performed through an open surgical approach and laparoscopic procedures are also performed.
Explain the Nursing management of the pyloric Stenosis.
Preoperative nursing management
To monitor the child’s vital signs regularly.
After providing the feeling to the child, provide him with an upright position and a little right side position.
Provide ADQ intravenous fluid to maintain child’s hydration status.
Properly maintain Child No Intake Output Chart.
Perform nasogastric drainage to empty the stomach.
Treatment procedure using isotonic solution.
Proper handling by maintaining aseptic technique to prevent infection of the child.
Maintain proper hand hygiene before handling the child.
Providing good skin care to the child.
Postoperative nursing management
To monitor the child’s vital signs regularly.
Provide adequate intravenous fluids to maintain the nutritional status of the child.
Maintain proper aseptic technique and provide dressing on the operative side.
Daily weight monitoring of child.
Continuously monitoring the child for any complications.
To provide education to the parents to provide adequate care of the child.
Providing complete education to the parents about the child’s condition.
Advising the child’s parents to provide the prescribed medication to the child.
To provide proper psychological support to the parents of the child.
Advising the child’s parents to follow up regularly.
Explain/Define Diaphragmatic Hernia
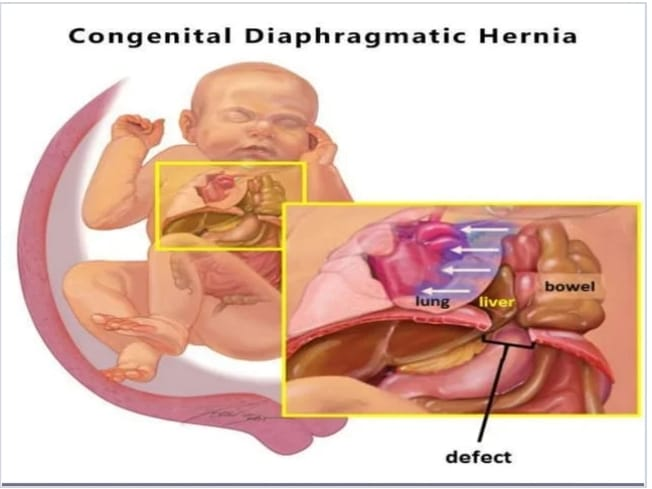
Hernia
Hernia is a condition in which any organ or part of an organ of the body protrudes or projects from its normal cavity through any wall or defect into another cavity, then the condition is called hernia.
Diaphragmatic hernia
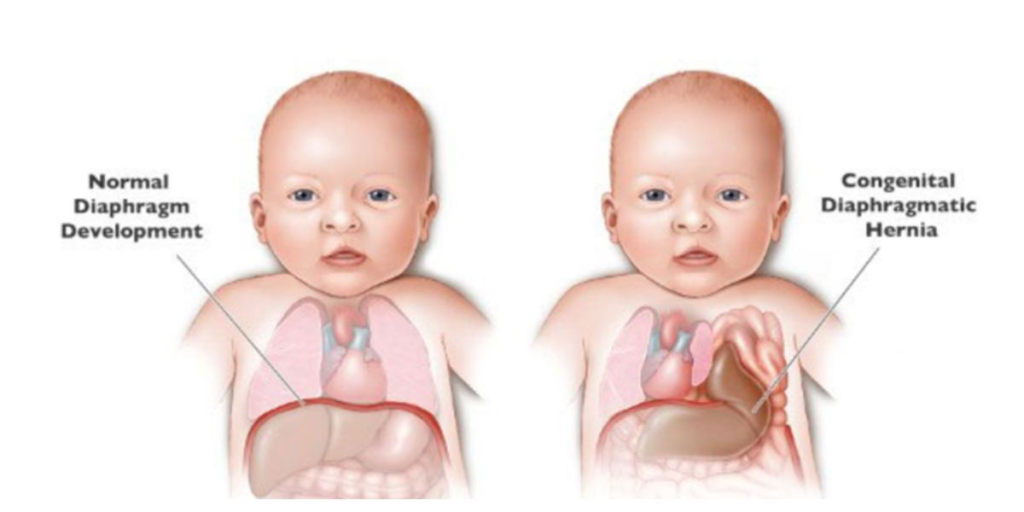
Diaphragmatic hernia is a congenital anomaly in children where there is a defect or abnormal opening in the diaphragm (the muscle that separates the chest cavity from the abdominal cavity.) This opening causes the organs of the abdominal cavity such as the stomach, spleen, intestines and The liver moves into the chest cavity. As a result, the affected lungs cannot develop properly due to the displaced abdominal organs, i.e. the lungs are compressed and collapsed. The severity of diaphragmatic hernia is different.
Explain the types of the congenital Diaphragmatic Hernia.
There are mainly two types of congenital diaphragmatic hernia.
Bochdalek hernia,
Morgagni hernia.
Bochdalek hernia
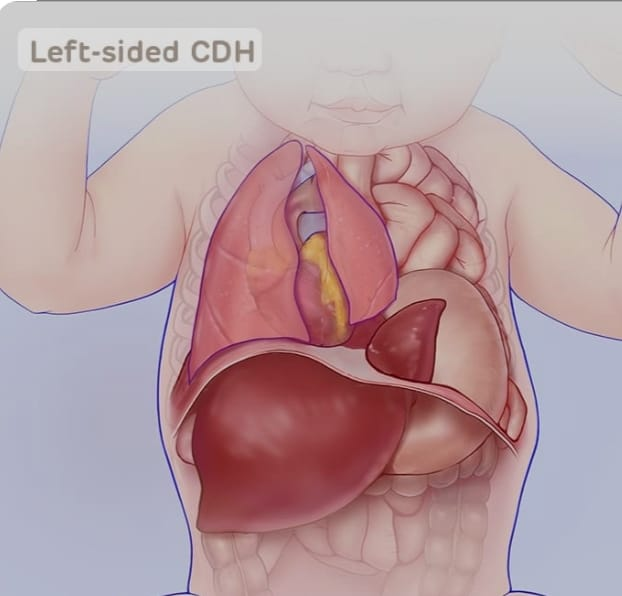
Bochdalek hernia is the most common type of congenital diaphragmatic hernia.
This type of hernia is usually seen due to a defective opening in the diaphragm on the left side of the diaphragm. Due to this opening, intestines and stomach protrude into the chest cavity.
Morgagni hernia.
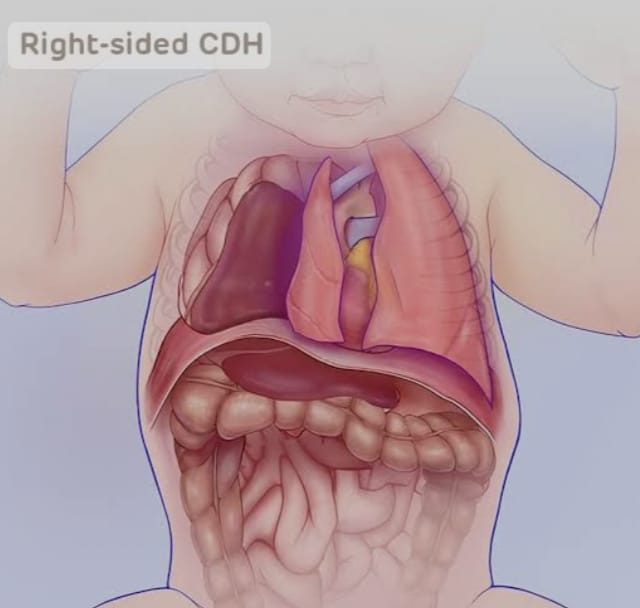
Morgagni hernia is a type of congenital diaphragmatic hernia.
This type of hernia is usually seen due to a defective opening in the diaphragm on the right side of the diaphragm. Due to this opening, intestines and liver protrude into the chest cavity.
Explain the Etiology/cause of the congenital Diaphragmatic Hernia
- The exact cause is unknown.
- Due to non-proper development of diaphragm during development time.
- Due to genetic factor.
- Due to environmental factors.
- Due to nutritional factors.
- Congenital.
Explain the Clinical manifestation/ sign and symptoms of the congenital Diaphragmatic Hernia.
- having dyspnoea,
- Respiratory distress after birth,
- rapid breeding,
- tachypnea,
- cyanosis,
- Chest size increases and abdomen becomes scaphoid (anterior abdominal wall becomes concave),
- Reduced chest movement and reduced breath sounds on the affected side.
- A peristalsis movement is heard in the chest of the affected side,
- Difficulty in fitting,
- vomiting,
- Failure to Thrive,
- grief,
- hypoxia.
Explain the Diagnostic evaluation of the congenital Diaphragmatic Hernia
- History taking and physical examination,
- prenatal screening,
- ultrasound,
- imaging studies,
- chest x-ray,
- laboratory test,
- blood test,
- Abdominal x-ray,
- Echocardiography.
Explain the Surgical management of the child with the congenital Diaphragmatic Hernia
Surgical management of congenital diaphragmatic hernia (CDH) usually involves a procedure called diaphragmatic hernia repair.
Surgical Repair:
Surgical repair is usually performed within the first few days to a few weeks of life, depending on the child’s stability.
The aim of the surgical procedure is to return the abdominal organs to their proper position in the abdomen and close the diaphragm defect. The procedure can be done through an open surgical approach or through minimally invasive techniques such as laparoscopy or thoracoscopy.
The surgeon carefully closes the hole in the diaphragm using sutures or patches.
Explain the Nursing management of the child with the congenital Diaphragmatic Hernia
Preoperative Nursing Management
Providing the child with a properly upright position.
Provide oxygen to the child using an endotracheal tube.
Properly draining respiratory secretions to prevent the child from infection.
Monitoring the child’s blood oxygen level regularly.
Decompress the stomach by aspiration through the nasogastric tube.
Continuous monitoring of child’s blood glucose level.
Provide intravenous fluids to maintain the nutritional status of the child.
Correct the condition of acidosis properly by providing sodium bicarbonate to the child.
Keep the child properly clothed to prevent hypothermia.
Provide proper antibiotic medicine to prevent the child from infection.
Postoperative nursing management
To continuously monitor the child’s vital signs.
Provide adequate intravenous fluids to maintain the nutritional status of the child.
Provide adequate respiratory support to the child.
Administer oxygen properly to the child.
Proper ventilation to keep the child’s air passage clear.
Maintaining body temperature of child continuously Avoiding exposure of child to external environment.
Providing a nutritious diet to the child.
Maintain proper hygienic condition to prevent child from infection.
Maintain proper aseptic technique and provide dressing on the operative side.
Daily weight monitoring of child.
Continuously monitoring the child for any complications.
To provide education to the parents to provide adequate care of the child.
Providing complete education to the parents about the child’s condition.
Advising the child’s parents to provide the prescribed medication to the child.
To provide proper psychological support to the parents of the child.
Advising the child’s parents to follow up regularly.
- Explain/Define Omphalocele (Exomphalus) in children Define Omphalocele (Exomphalus).
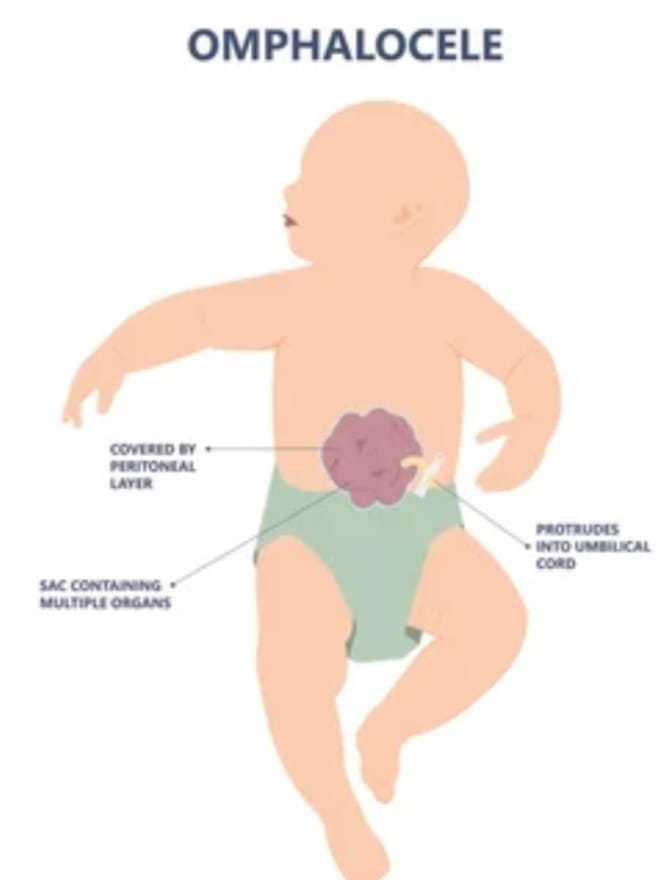
Omphalocele, also known as exomphallus, is a congenital abdominal wall defect that occurs during fetal development.
In an omphalocele, organs of the abdomen, such as the intestines, liver, and other abdominal organs protrude (herniate) outside the body from the base of the umbilical cord. The omphalocele forms a sac covered by a layer of peritoneum and a layer of amniotic membrane that herniates from the umbilicus into a sac-like structure outside the body.
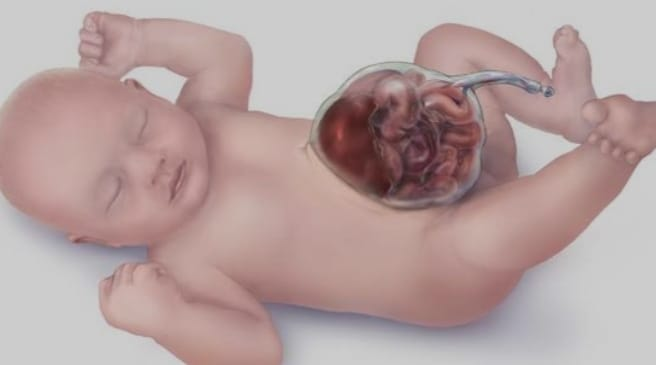
Omphalocele differs in severity, with some cases involving only a small portion of the herniated tissue while others may involve many organs of the abdomen.
Surgical correction is required to treat it.
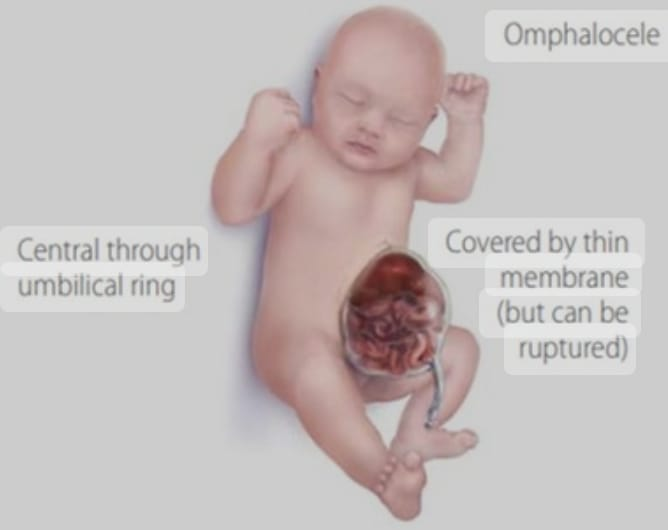
Explain the Etiology/cause of the child with the Omphalocele.
Due to
genetic factors,
Due to chromosomal abnormalities like trisomy 13, trisomy 18, trisomy 21.
Beckwith-Wiedemann syndrome (omphalocele, macroglossia, macrosomia, microcephaly)
Due to alcohol and smoking intake by the mother during pregnancy.
Due to the mother taking certain medications during pregnancy such as Selective Serotonin Reuptake Inhibitors (SSRI),
Due to carrying extra weight by mother during pregnancy time.
Due to any type of infection in the mother during pregnancy.
Due to certain maternal factors like maternal age, obesity, diabetes etc.,
Due to abdominal wall not developing properly during fetal development time.
Explain the Clinical manifestation/ sign and symptoms of the child with the Omphalocele.
- Visible abdominal swelling,
- A sac like structure visible at the abdominal site.
- Respiratory distress.
- Difficulty in feeding.
- Other congenital anomalies are also seen with omphalocele such as,
- cardiac defect,
- Genitourinary Abnormalities,
- Chromosomal Abnormalities etc.,
- Other complications are also seen like,
- Rupture of sac,
- getting an infection,
- Intestinal obstruction,
- hypothermia,
- hypovolemia,
- Getting sepsis.
Explain the Diagnostic evaluation of the child with the Omphalocele.
- History taking and physical examination,
- Prenatal Second and Third Trimester Ultrasound,
- imaging studies,
- Laboratory test,
- blood test,
- Complete blood count test,
Explain the Surgical management of the child with the Omphalocele.
Surgical repair
Surgical repair of omphalocele usually involves stages.
In the first stage,
The herniated organ is carefully reinserted into the abdominal cavity. If the defect is large or there is inadequate tissue to close the abdominal cavity, a temporary covering, such as silos or synthetic materials, is used.
Second stage
Once the herniated organ is inserted back into the abdominal cavity, the abdominal wall defect is closed using surgical techniques.
Providing antibiotic medication to prevent the child from infection.
Explain the Nursing management of the child with the Omphalocele.
Preoperative nursing management
Conducting a properly comparative assessment of the child.
To properly assess the child’s vital signs.
To properly assess the child’s respiratory status.
Properly assess the nutritional status of the child.
Properly stabilizing the child like providing adequate oxygen, providing proper ventilatory support.
Provide proper medication to the child.
Providing proper nutritional support to the child.
Maintain proper aseptic technique and provide dressing to prevent infection of omphalosil sac to the child.
To provide proper emotional and psychological support to the family members of the child.
Post operative management
Properly assess the child’s pain level.
If the child is in pain, provide proper analgesic medication.
Provide a properly comfortable position to the child.
Properly assess the child’s respiratory status.
Provide oxygen properly to improve the child’s respiratory condition.
Provide proper ventilatory support to the child.
Properly assess the child’s surgical site for any type of infection and drainage.
Maintain proper aseptic technique and provide dressings to improve incision site healing and prevent infection.
Providing proper nutritional support to the child.
Proper assessment of the child for any type of complications like infection, respiratory distress, gastro-intestinal infection or not.
To provide complete education to the child and his family members about the child’s condition, its causes, symptoms and signs, and its treatment.
To provide proper psychological support and counseling to the family members of the child.
- Explain/Define Imperforated Anus.
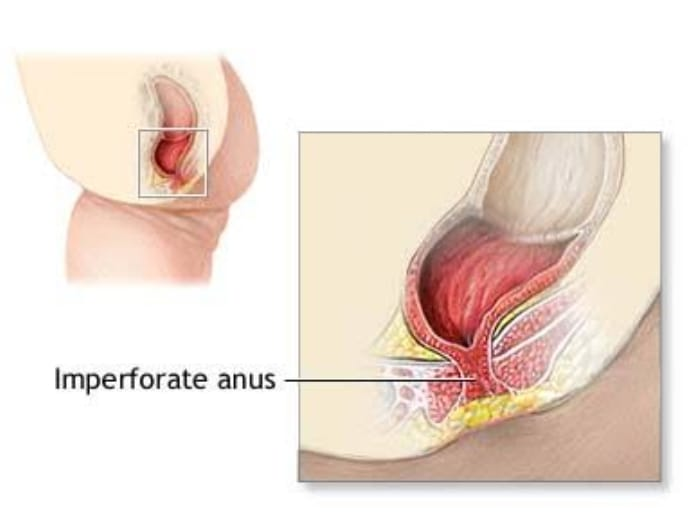
An imperforate anus, also known as anus atresia, is a congenital condition in which the opening of the anus is absent or
Improperly formed. This means that there is no opening for stool to pass from the rectum to the outside of the body. Severity of imperforate anus varies, ranging from involvement of the membrane covering the anal opening to complete absence of the anus.
Explain the types of Imperforated Anus
There are five types of imperfect anus.
1) Anal stenosis,
2) Anal membrane,
3) Low imperforate anus,
4) High imperforate anus,
5) Rectal atresia
••>
1) Anal stenosis,
Anal stenosis is a condition in which there is narrowing and tightening of the anal canal causing difficulty in passing stool.
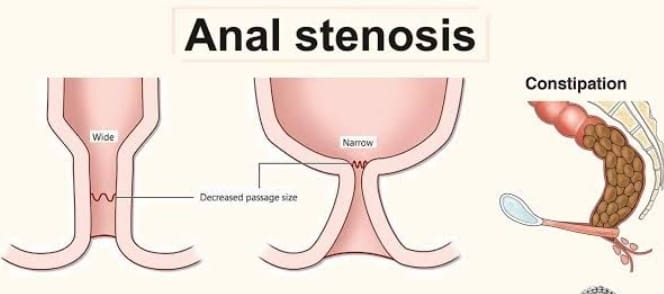
2) Anal membrane,
Anal membrane is a congenital condition in which there is obstruction by thin membrane or tissues at the site of the anal opening. This obstruction can be complete or partial as this type of membrane usually forms during fetal development which causes difficulty in passing stool.
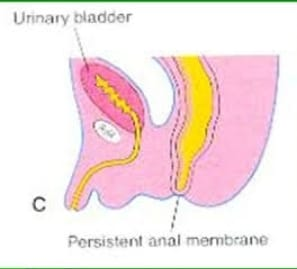
3) Low imperforate anus,
In this type the distance between the blind pouch of the rectum and the anal dimple is less than 1.5 cm, the rectum is descended below the pubococcygeal line.
4) High imperforate anus,
In this type, the distance between the blind pouch of the rectum and the anal dimple is more than 1.5 cm, the rectal pouch is present above the pubococcygeal line.
5) Rectal atresia
It is a congenital condition in which the rectum is absent.
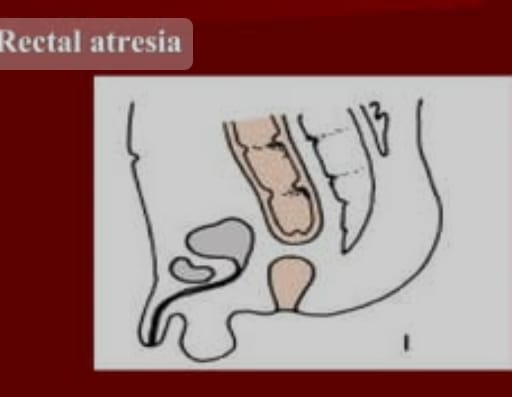
Explain the Clinical manifestation/ sign and symptoms of the child with the Imperforated Anus.
- Absent anal opening,
- Failure to pass meconium during 24 to 48 hours after birth of the newborn.
- Abdominal distension.
- Swelling in the abdomen due to stool buildup in the intestines.
- Vomiting.
- Failure to thrive.
- Irritability.
- Discomfort.
Explain the Diagnostic evaluation of the child with the Imperforated Anus.
- History taking and physical examination,
- imaging studies,
- ultrasound,
- x ray,
- Lower Gastrointestinal Track MRI
- Rectal examination.
- Genitourinary evaluation.
- Blood test.
- Inspection of anal opening.
Explain the Surgical management of the child with the Imperforated Anus.
Anal stenosis
Anal stenosis is manually dilated.
Low imperforate anus
Anoplasty of low imperforate anus is usually performed during the neonatal period and early infancy period.
High imperforated anus
In highly imperforated anus, colestomy is performed during late infancy and when weight is 8 kg.
Explain the Nursing management of the child with the Imperforated Anus.
Preoperative nursing management
Proper inspection of new born.
Assessing whether the newborn passes meconium at 24 to 48 hours.
After confirming the diagnosis, providing information about the child’s condition to the parents of the child properly.
To provide complete education to the child’s parents about the child’s condition, its signs and symptoms and its treatment and surgical procedures.
To provide proper reassurance to parents to accept their child.
Keep the child on NPO.
Provide intravenous fluids to maintain the nutritional status of the child.
Properly measuring the child’s abdominal girth.
Perform nasogastric aspiration and perform gastric decompression.
Properly maintain the child’s body temperature.
To monitor the child’s vital signs regularly.
Postoperative nursing management
Provide proper postanesthetic care to the child.
Maintain proper aseptic technique to prevent child from infection.
To monitor the child’s vital signs properly.
Keeping the stool performed in the child’s body properly clean and maintaining its hygienic condition.
Properly observe the circulation on the side where the stoma is done and the color of that site.
Stow site should be kept properly clean by maintaining aseptic technique and covered by proper dressing.
To observe the urine output of the child.
Assess the child for any other complications such as abdominal distension, bleeding conditions or not.
Properly monitoring the child’s fluid and electrolyte levels.
Provide intravenous fluids to properly maintain the nutritional status of the child.
Provide proper side lining position to the child and raise his legs.
Parental Advice
To provide complete education to the child’s parents about the child’s condition.
To provide education to the parents of the child to take care of the colostomy performed on the child and to give advice to the parents to provide the care of the colostomy by demonstrating it.
Advise the child’s parents to perform regular anal dilatation to maintain anal opening.
Advise the parents of the child to provide toilet training to the child at regular time.
Provide complete education to parents about child’s medication, hygienic condition, and diet.
To provide complete education to the parents if the child has to perform any surgery in the future.
Advise parents to maintain proper hygienic condition of child to prevent infection.
Advise parents to keep child’s anal area properly dry and clean.
Advising parents to provide properly prescribed medication to their child.
Advising parents to follow up regularly.